Dosen : Dr. rer. nat. I Made Wiryana, SKom, SSi, MAppSc
Julia Christie Feliciana De Chantal
KATA PENGANTAR
Puji dan syukur kami panjatkan kepada Tuhan Yang Maha Esa karena atas berkat rahmat dan karunia-Nya, serta dorongan doa restu, dan dorongan dari berbagai pihak sehingga kelompok kami dapat menyelesaikan tugas penulisan buku ini dengan judul “BIOINFORMATIC”. Kami dari tim penulis mengucapkan banyak terimakasih kepada Bapak I Made Wiryana S. Kom. MApp Sc yang telah memberikan bimbingan maupun arahan kepada kelompok kami sehingga kami bisa memahami tentang tugas yang telah diberikan oleh bapak tersebut. Terima kasih juga kepada teman-teman kelas 4IA08 yang telah turut membantu dalam memberikan informasi seputar cara pengerjaan tugas ini. Kami dari tim penulis menyadari bahwa buku yang kami tulis ini masih jauh dari kata sempurna, sehingga jika ada penulisan nama maupun materi yang salah harap dimaklumi. Besar harapan kami dari tim penulis dengan adanya buku ini kami berharap dapat membantu sekaligus memberikan informasi kepada pembaca agar dapat dimanfaatkan oleh pembaca di kemudian hari.
BAB 1
PENDAHULUAN
Dengan semakin berkembangnya zaman, semakin pula berkembangnya sebuah teknologi. Banyak penemu-penemu baru bermunculan dan juga pengembangan dari teknologi yang sudah ada sebelumnya yang memiliki dampak positif bagi kehidupan manusia. Salah satunya yaitu teknologi dibidang Bioinformatika atau dalam Bahasa inggris bioinformatics.
Bioinformatika adalah ilmu yang mempelajari atau sebuah penerapan teknik komputasional untuk mengelola dan menganalisis informasi biologis. Bidang ini mencakup penerapan metode-metode matematika, statistika, dan informatika untuk memecahkan masalah-masalah biologis, terutama dengan menggunakan sekuens DNA dan asam amino serta informasi yang berkaitan dengannya. Contoh topik utama bidang ini meliputi basis data untuk mengelola informasi biologis, penyejajaran sekuens (sequence alignment), prediksi struktur untuk meramalkan bentuk struktur protein maupun struktur sekunder RNA, analisis filogenetik, dan analisis ekspresi gen.
Berbagai kasus mulai dari berbagai penelitian gen, persilangan kromosom pada hewan dan tumbuhan dan teknologi lain yang cukup besar untuk menghasilkan sesuatu yang baru yang merupakan tantangan bagi para spesialis komputerisasi untuk mengembangkan ilmunya dengan selalu memperbaharui atau menambah wawasan yang dimilikinya sehingga terciptalah suatu strategi penanganan yang lebih efektif dan efisien.
Buku ini membahas tentang BIOINFORMATIKA menggunakan software GENTLE perangkat lunak untuk mengedit DNA dan asam amino, manajemen database, peta plasmid, pembatasan dan ligasi, keberpihakan, sequencer data impor, kalkulator, tampilan gambar gel, PCR, dan banyak lagi. Ini adalah perangkat lunak gratis di bawah GPLv2 dan dapat diunduh secara gratis di
http://gentle.magnusmanske.de/
BAB 2
LANDASAN TEORI
2.1 Bioinformatika
2.1.1 Pengertian
Bioinformatika adalah ilmu yang mempelajari penerapan teknikkomputasional untuk mengelola dan menganalisis informasi biologis. Bidang ini mencakup penerapan metode-metode matematika, statistika, dan informatika untuk memecahkan masalah-masalah biologis, terutama dengan menggunakan sekuens DNA dan asam amino serta informasi yang berkaitan dengannya. Contoh topik utama bidang ini meliputi basis data untuk mengelola informasi biologis, penyejajaran sekuens (sequence alignment), prediksi struktur untuk meramalkan bentuk struktur protein maupun struktur sekunder RNA, analisis filogenetik, dan analisis ekspresi gen.
2.1.2 Sejarah
Istilah bioinformatics mulai dikemukakan pada pertengahan era 1980-an untuk mengacu pada penerapan komputer dalam biologi. Namun demikian, penerapan bidang-bidang dalam bioinformatika (seperti pembuatan basis data dan pengembangan algoritma untuk analisis sekuens biologis) sudah dilakukan sejak tahun 1960-an.
Kemajuan teknik biologi molekular dalam mengungkap sekuens biologis dari protein (sejak awal 1950-an) dan asam nukleat (sejak 1960-an) mengawali perkembangan basis data dan teknik analisis sekuens biologis. Basis data sekuens protein mulai dikembangkan pada tahun 1960-an di Amerika Serikat, sementara basis data sekuens DNA dikembangkan pada akhir 1970-an di Amerika Serikat dan Jerman (pada European Molecular Biology Laboratory, Laboratorium Biologi Molekular Eropa). Penemuan teknik sekuensing DNA yang lebih cepat pada pertengahan 1970-an menjadi landasan terjadinya ledakan jumlah sekuens DNA yang berhasil diungkapkan pada 1980-an dan 1990-an, menjadi salah satu pembuka jalan bagi proyek-proyek pengungkapan genom, meningkatkan kebutuhan akan pengelolaan dan analisis sekuens, dan pada akhirnya menyebabkan lahirnya bioinformatika.
Perkembangan Internet juga mendukung berkembangnya bioinformatika. Basis data bioinformatika yang terhubung melalui Internet memudahkan ilmuwan mengumpulkan hasil sekuensing ke dalam basis data tersebut maupun memperoleh sekuens biologis sebagai bahan analisis. Selain itu, penyebaran program-program aplikasi bioinformatika melalui Internet memudahkan ilmuwan mengakses program-program tersebut dan kemudian memudahkan pengembangannya.
2.1.3 Penerapan Bioinformatika
2.1.3.1 Basis Data Sekuens Biologis
Sesuai dengan jenis informasi biologis yang disimpannya, basis data sekuens biologis dapat berupa basis data primer untuk menyimpan sekuens primer asam nukleat maupunprotein, basis data sekunder untuk menyimpan motif sekuens protein, dan basis data struktur untuk menyimpan data struktur protein maupun asam nukleat.
Basis data utama untuk sekuens asam nukleat saat ini adalah GenBank (Amerika Serikat), EMBL (Eropa), dan DDBJ (Inggris) (DNA Data Bank of Japan, Jepang). Ketiga basis data tersebut bekerja sama dan bertukar data secara harian untuk menjaga keluasan cakupan masing-masing basis data. Sumber utama data sekuens asam nukleat adalah submisi langsung dari periset individual, proyek sekuensing genom, dan pendaftaran paten. Selain berisi sekuens asam nukleat, entri dalam basis data sekuens asam nukleat umumnya mengandung informasi tentang jenis asam nukleat (DNA atau RNA), nama organisme sumber asam nukleat tersebut, dan pustaka yang berkaitan dengan sekuens asam nukleat tersebut.
Sementara itu, contoh beberapa basis data penting yang menyimpan sekuens primer protein adalah PIR (Protein Information Resource, Amerika Serikat), Swiss-Prot (Eropa), dan TrEMBL (Eropa). Ketiga basis data tersebut telah digabungkan dalam UniProt (yang didanai terutama oleh Amerika Serikat). Entri dalam UniProt mengandung informasi tentang sekuens protein, nama organisme sumber protein, pustaka yang berkaitan, dan komentar yang umumnya berisi penjelasan mengenai fungsi protein tersebut.
BLAST (Basic Local Alignment Search Tool) merupakan perkakas bioinformatika yang berkaitan erat dengan penggunaan basis data sekuens biologis. Penelusuran BLAST (BLAST search) pada basis data sekuens memungkinkan ilmuwan untuk mencari sekuens asam nukleat maupun protein yang mirip dengan sekuens tertentu yang dimilikinya. Hal ini berguna misalnya untuk menemukan gen sejenis pada beberapa organisme atau untuk memeriksa keabsahan hasil sekuensing maupun untuk memeriksa fungsi gen hasil sekuensing. Algoritma yang mendasari kerja BLAST adalah penyejajaran sekuens.
PDB (Protein Data Bank, Bank Data Protein) adalah basis data tunggal yang menyimpan model struktural tiga dimensi protein dan asam nukleat hasil penentuan eksperimental (dengan kristalografi sinar-X, spektroskopi NMR dan mikroskopi elektron). PDB menyimpan data struktur sebagai koordinat tiga dimensi yang menggambarkan posisi atom-atom dalam protein ataupun asam nukleat.
2.1.3.2 Penyejajaran Sekuens
Penyejajaran sekuens (sequence alignment) adalah proses penyusunan/pengaturan dua atau lebih sekuens sehingga persamaan sekuens-sekuens tersebut tampak nyata. Hasil dari proses tersebut juga disebut sebagai sequence alignment atau alignment saja. Baris sekuens dalam suatu alignment diberi sisipan (umumnya dengan tanda "-") sedemikian rupa sehingga kolom-kolomnya memuat karakter yang identik atau sama di antara sekuens-sekuens tersebut. Berikut adalah contoh alignment DNA dari dua sekuens pendek DNA yang berbeda, "ccatcaac" dan "caatgggcaac" (tanda "|" menunjukkan kecocokan atau match di antara kedua sekuens).
Sequence alignment merupakan metode dasar dalam analisis sekuens. Metode ini digunakan untuk mempelajari evolusi sekuens-sekuens dari leluhur yang sama (common ancestor). Ketidakcocokan (mismatch) dalam alignment diasosiasikan dengan proses mutasi, sedangkan kesenjangan (gap, tanda "-") diasosiasikan dengan proses insersi atau delesi. Sequence alignment memberikan hipotesis atas proses evolusi yang terjadi dalam sekuens-sekuens tersebut. Misalnya, kedua sekuens dalam contoh alignment di atas bisa jadi berevolusi dari sekuens yang sama "ccatgggcaac". Dalam kaitannya dengan hal ini, alignment juga dapat menunjukkan posisi-posisi yang dipertahankan (conserved) selama evolusi dalam sekuens-sekuens protein, yang menunjukkan bahwa posisi-posisi tersebut bisa jadi penting bagi struktur atau fungsi protein tersebut.
Selain itu, sequence alignment juga digunakan untuk mencari sekuens yang mirip atau sama dalam basis data sekuens. BLAST adalah salah satu metode alignment yang sering digunakan dalam penelusuran basis data sekuens. BLAST menggunakan algoritma heuristik dalam penyusunan alignment.
Beberapa metode alignment lain yang merupakan pendahulu BLAST adalah metode "Needleman-Wunsch" dan "Smith-Waterman". Metode Needleman-Wunsch digunakan untuk menyusun alignment global di antara dua atau lebih sekuens, yaitu alignment atas keseluruhan panjang sekuens tersebut. Metode Smith-Waterman menghasilkan alignmentlokal, yaitu alignment atas bagian-bagian dalam sekuens. Kedua metode tersebut menerapkan pemrograman dinamik (dynamic programming) dan hanya efektif untuk alignmentdua sekuens (pairwise alignment)
Clustal adalah program bioinformatika untuk alignment multipel (multiple alignment), yaitu alignment beberapa sekuens sekaligus. Dua varian utama Clustal adalah ClustalWdan ClustalX.
Metode lain yang dapat diterapkan untuk alignment sekuens adalah metode yang berhubungan dengan Hidden Markov Model ("Model Markov Tersembunyi", HMM). HMM merupakan model statistika yang mulanya digunakan dalam ilmu komputer untuk mengenali pembicaraan manusia (speech recognition). Selain digunakan untuk alignment, HMM juga digunakan dalam metode-metode analisis sekuens lainnya, seperti prediksi daerah pengkode protein dalam genom dan prediksi struktur sekunder protein.
2.1.3.3 Prediksi struktur protein
Secara kimia/fisika, bentuk struktur protein diungkap dengan kristalografi sinar-X ataupun spektroskopi NMR, namun kedua metode tersebut sangat memakan waktu dan relatif mahal. Sementara itu, metode sekuensing protein relatif lebih mudah mengungkapkan sekuensasam amino protein. Prediksi struktur protein berusaha meramalkan struktur tiga dimensi protein berdasarkan sekuens asam aminonya (dengan kata lain, meramalkan struktur tersier dan struktur sekunder berdasarkan struktur primer protein). Secara umum, metode prediksi struktur protein yang ada saat ini dapat dikategorikan ke dalam dua kelompok, yaitu metode pemodelan protein komparatif dan metode pemodelan de novo.
Pemodelan protein komparatif (comparative protein modelling) meramalkan struktur suatu protein berdasarkan struktur protein lain yang sudah diketahui. Salah satu penerapan metode ini adalah pemodelan homologi (homology modelling), yaitu prediksi struktur tersier protein berdasarkan kesamaan struktur primer protein. Pemodelan homologi didasarkan pada teori bahwa dua protein yanghomolog memiliki struktur yang sangat mirip satu sama lain. Pada metode ini, struktur suatu protein (disebut protein target) ditentukan berdasarkan struktur protein lain (protein templat) yang sudah diketahui dan memiliki kemiripan sekuens dengan protein target tersebut. Selain itu, penerapan lain pemodelan komparatif adalah protein threading yang didasarkan pada kemiripan struktur tanpa kemiripan sekuens primer. Latar belakang protein threading adalah bahwa struktur protein lebih dikonservasi daripada sekuens protein selama evolusi; daerah-daerah yang penting bagi fungsi protein dipertahankan strukturnya. Pada pendekatan ini, struktur yang paling kompatibel untuk suatu sekuens asam amino dipilih dari semua jenis struktur tiga dimensi protein yang ada. Metode-metode yang tergolong dalamprotein threading berusaha menentukan tingkat kompatibilitas tersebut.
Dalam pendekatan de novo atau ab initio, struktur protein ditentukan dari sekuens primernya tanpa membandingkan dengan struktur protein lain. Terdapat banyak kemungkinan dalam pendekatan ini, misalnya dengan menirukan proses pelipatan (folding) protein dari sekuens primernya menjadi struktur tersiernya (misalnya dengan simulasi dinamika molekular), atau dengan optimisasi global fungsi energi protein. Prosedur-prosedur ini cenderung membutuhkan proses komputasi yang intens, sehingga saat ini hanya digunakan dalam menentukan struktur protein-protein kecil. Beberapa usaha telah dilakukan untuk mengatasi kekurangan sumber daya komputasi tersebut, misalnya dengan superkomputer (misalnya superkomputer Blue Gene dari IBM) atau komputasi terdistribusi maupun komputasi grid.
2.1.3.4 Analisis ekspresi gen
Ekspresi gen dapat ditentukan dengan mengukur kadar mRNA dengan berbagai macam teknik (misalnya dengan microarray ataupunSerial Analysis of Gene Expression ["Analisis Serial Ekspresi Gen", SAGE]). Teknik-teknik tersebut umumnya diterapkan pada analisis ekspresi gen skala besar yang mengukur ekspresi banyak gen (bahkan genom) dan menghasilkan data skala besar. Metode-metode penggalian data (data mining) diterapkan pada data tersebut untuk memperoleh pola-pola informatif. Sebagai contoh, metode-metode komparasi digunakan untuk membandingkan ekspresi di antara gen-gen, sementara metode-metode klastering (clustering) digunakan untuk mempartisi data tersebut berdasarkan kesamaan ekspresi gen.
2.1.4 Contoh-contoh Penggunaan
2.1.4.1 Bioinformatika dalam Bidang Klinis
Bioinformatika dalam bidang klinis sering disebut sebagai informatika klinis (clinical informatics). Aplikasi dari informatika klinis ini berbentuk manajemen data-data klinis dari pasien melalui Electrical Medical Record (EMR) yang dikembangkan oleh Clement J. McDonald dari Indiana University School of Medicine pada tahun 1972. McDonald pertama kali mengaplikasikan EMR pada 33 orang pasien penyakit gula (diabetes). Sekarang EMR ini telah diaplikasikan pada berbagai penyakit. Data yang disimpan meliputi data analisa diagnosa laboratorium, hasil konsultasi dan saran, foto rontgen, ukuran detak jantung, dan lain lain. Dengan data ini dokter akan bisa menentukan obat yang sesuai dengan kondisi pasien tertentu dan lebih jauh lagi, dengan dibacanya genom manusia, akan memungkinkan untuk mengetahui penyakit genetik seseorang, sehingga penanganan terhadap pasien menjadi lebih akurat.
2.1.4.2 Bioinformatika untuk Identifikasi Agent Penyakit Baru
Bioinformatika juga menyediakan tool yang sangat penting untuk identifikasi agent penyakit yang belum dikenal penyebabnya. Banyak sekali penyakit baru yang muncul dalam dekade ini, dan diantaranya yang masih hangat adalah SARS (Severe Acute Respiratory Syndrome). Pada awalnya, penyakit ini diperkirakan disebabkan oleh virus influenza karena gejalanya mirip dengan gejala pengidap influenza. Akan tetapi ternyata dugaan ini salah karena virus influenza tidak terisolasi dari pasien. Perkirakan lain penyakit ini disebabkan oleh bakteri Candida karena bakteri ini terisolasi dari beberapa pasien. Tapi perkiraan ini juga salah. Akhirnya ditemukan bahwa dari sebagian besar pasien SARS terisolasi virus Corona jika dilihat dari morfologinya.
Sekuen genom virus ini kemudian dibaca dan dari hasil analisa dikonfirmasikan bahwa penyebab SARS adalah virus Corona yang telah berubah (mutasi) dari virus Corona yang ada selama ini. Dalam rentetan proses ini, Bioinformatika memegang peranan penting. Pertama pada proses pembacaan genom virus Corona. Karena di database seperti GenBank, EMBL (European Molecular Biology Laboratory), dan DDBJ (DNA Data Bank of Japan) sudah tersedia data sekuen beberapa virus Corona, yang bisa digunakan untuk mendisain primer yang digunakan untuk amplifikasi DNA virus SARS ini. Software untuk mendisain primer juga tersedia, baik yang gratis maupun yang komersial. Contoh yang gratis adalah Webprimer yang disediakan oleh Stanford Genomic Resources (http://genome-www2.stanford.edu/cgi-bin/SGD/web-primer), GeneWalker yang disediakan oleh Cybergene AB (http://www.cybergene.se/primerdisain/genewalker), dan lain sebagainya. Untuk yang komersial ada Primer Disainer yang dikembangkan oleh Scientific & Education Software, dan software-software untuk analisa DNA lainnya seperti Sequencher (GeneCodes Corp.), SeqMan II (DNA STAR Inc.), Genetyx (GENETYX Corp.), DNASIS (HITACHI Software), dan lain lain.
Kedua pada proses mencari kemiripan sekuen (homology alignment) virus yang didapatkan dengan virus lainnya. Dari hasil analisa virus SARS diketahui bahwa genom virus Corona penyebab SARS berbeda dengan virus Corona lainnya. Perbedaan ini diketahui dengan menggunakan homology alignment dari sekuen virus SARS. Selanjutnya, Bioinformatika juga berfungsi untuk analisa posisi sejauh mana suatu virus berbeda dengan virus lainnya.
2.1.4.3 Bioinformatika untuk Diagnosa Penyakit Baru
Untuk menangani penyakit baru diperlukan diagnosa yang akurat sehingga dapat dibedakan dengan penyakit lain. Diagnosa yang akurat ini sangat diperlukan untuk pemberian obat dan perawatan yang tepat bagi pasien. Ada beberapa cara untuk mendiagnosa suatu penyakit, antara lain: isolasi agent penyebab penyakit tersebut dan analisa morfologinya, deteksi antibodi yang dihasilkan dari infeksi dengan teknik enzyme-linked immunosorbent assay (ELISA), dan deteksi gen dari agent pembawa penyakit tersebut dengan Polymerase Chain Reaction (PCR).
Teknik yang banyak dan lazim dipakai saat ini adalah teknik PCR. Teknik ini sederhana, praktis dan cepat. Yang penting dalam teknik PCR adalah disain primer untuk amplifikasi DNA, yang memerlukan data sekuen dari genom agent yang bersangkutan dan software seperti yang telah diuraikan di atas. Disinilah Bioinformatika memainkan peranannya. Untuk agent yang mempunyai genom RNA, harus dilakukan reverse transcription (proses sintesa DNA dari RNA) terlebih dahulu dengan menggunakan enzim reverse transcriptase.
Setelah DNA diperoleh baru dilakukan PCR. Reverse transcription dan PCR ini bisa dilakukan sekaligus dan biasanya dinamakan RT-PCR. Teknik PCR ini bersifat kualitatif, oleh sebab itu sejak beberapa tahun yang lalu dikembangkan teknik lain, yaitu Real Time PCR yang bersifat kuantitatif. Dari hasil Real Time PCR ini bisa ditentukan kuantitas suatu agent di dalam tubuh seseorang, sehingga bisa dievaluasi tingkat emergensinya. Pada Real Time PCR ini selain primer diperlukan probe yang harus didisain sesuai dengan sekuen agent yang bersangkutan. Di sini juga diperlukan software atau program Bioinformatika.
2.1.4.4 Bioinformatika untuk Penemuan Obat
Cara untuk menemukan obat biasanya dilakukan dengan menemukan zat/senyawa yang dapat menekan perkembangbiakan suatu agent penyebab penyakit. Karena perkembangbiakan agent tersebut dipengaruhi oleh banyak faktor, maka faktor-faktor inilah yang dijadikan target. Diantaranya adalah enzim-enzim yang diperlukan untuk perkembangbiakan suatu agent Mula-mula yang harus dilakukan adalah analisa struktur dan fungsi enzim-enzim tersebut. Kemudian mencari atau mensintesa zat/senyawa yang dapat menekan fungsi dari enzim-enzim tersebut. Analisa struktur dan fungsi enzim ini dilakukan dengan cara mengganti asam amino tertentu dan menguji efeknya. Analisa penggantian asam amino ini dahulu dilakukan secara random sehingga memerlukan waktu yang lama.
Setelah Bioinformatika berkembang, data-data protein yang sudah dianalisa bebas diakses oleh siapapun, baik data sekuen asam amino-nya seperti yang ada di SWISS-PROT (http://www.ebi.ac.uk/swissprot/) maupun struktur 3D-nya yang tersedia di Protein Data Bank (PDB) (http://www.rcsb.org/pdb/). Dengan database yang tersedia ini, enzim yang baru ditemukan dapat dibandingkan sekuen asam amino-nya, sehingga bisa diperkirakan asam amino yang berperan untuk aktivitas (active site) dan kestabilan enzim tersebut. Setelah asam amino yang berperan sebagai active site dan kestabilan enzim tersebut ditemukan, kemudian dicari atau disintesa senyawa yang dapat berinteraksi dengan asam amino tersebut.
Dengan data yang ada di PDB, maka dapat dilihat struktur 3D suatu enzim termasuk active site-nya, sehingga bisa diperkirakan bentuk senyawa yang akan berinteraksi dengan active site tersebut. Dengan demikian, kita cukup mensintesa senyawa yang diperkirakan akan berinteraksi, sehingga obat terhadap suatu penyakit akan jauh lebih cepat ditemukan. Cara ini dinamakan “docking” dan telah banyak digunakan oleh perusahaan farmasi untuk penemuan obat baru. Meskipun dengan Bioinformatika ini dapat diperkirakan senyawa yang berinteraksi dan menekan fungsi suatu enzim, namun hasilnya harus dikonfirmasi dahulu melalui eksperimen di laboratorium. Akan tetapi dengan Bioinformatika, semua proses ini bisa dilakukan lebih cepat sehingga lebih efisien baik dari segi waktu maupun finansial. Tahun 1997, Ian Wilmut dari Roslin Institute dan PPL Therapeutics Ltd, Edinburgh, Skotlandia, berhasil mengklon gen manusia yang menghasilkan faktor IX (faktor pembekuan darah), dan memasukkan ke kromosom biri-biri. Diharapkan biri-biri yang selnya mengandung gen manusia faktor IX akan menghasilkan susu yang mengandung faktor pembekuan darah. Jika berhasil diproduksi dalam jumlah banyak maka faktor IX yang diisolasi dari susu harganya bisa lebih murah untuk membantu para penderita hemofilia.
2.1.5 Pengertian Secara Khusus
Pada bagian pendahuluan kita telah diberikan gambaran sekilas mengenai perkembangan dan apa yang dapat diberikan oleh Bioinformatika. Bagian berikut ini akan membahas lebih detail tentang Bioinformatika. Secara umum, Bioinformatika dapat digambarkan sebagai: segala bentuk penggunaan komputer dalam menangani informasi-informasi biologi. Dalam prakteknya, definisi yang digunakan oleh kebanyakan orang bersifat lebih terperinci. Bioinformatika menurut kebanyakan orang adalah satu sinonim dari komputasi biologi molekul (penggunaan komputer dalam menandai karakterisasi dari komponenkomponen molekul dari makhluk hidup).
2.1.5.1 Bioinformatika "klasik"
Sebagian besar ahli Biologi mengistilahkan ‘mereka sedang melakukan Bioinformatika’ ketika mereka sedang menggunakan komputer untuk menyimpan, melihat atau mengambil data, menganalisa atau memprediksi komposisi atau struktur dari biomolekul. Ketika kemampuan komputer menjadi semakin tinggi maka proses yang dilakukan dalam Bioinformatika dapat ditambah dengan melakukan simulasi. Yang termasuk biomolekul diantaranya adalah materi genetik dari manusia --asam nukleat-- dan produk dari gen manusia, yaitu protein. Hal-hal diataslah yang merupakan bahasan utama dari Bioinformatika "klasik", terutama berurusan dengan analisis sekuen (sequence analysis). Definisi Bioinformatika menurut Fredj Tekaia dari Institut Pasteur [TEKAIA2004] adalah: "metode matematika, statistik dan komputasi yang bertujuan untuk menyelesaikan masalah-masalah biologi dengan menggunakan sekuen DNA dan asam amino dan informasi-informasi yang terkait dengannya."
Dari sudut pandang Matematika, sebagian besar molekul biologi mempunyai sifat yang menarik, yaitu molekul-molekul tersebut adalah polymer; rantai-rantai yang tersusun rapi dari modul-modul molekul yang lebih sederhana, yang disebut monomer. Monomer dapat dianalogikan sebagai bagian dari bangunan, dimana meskipun bagianbagian tersebut berbeda warna dan bentuk, namun semua memiliki ketebalan yang sama dan cara yang sama untuk dihubungkan antara yang satu dengan yang lain. Monomer yang dapat dikombinasi dalam satu rantai ada dalam satu kelas umum yang sama, namun tiap jenis monomer dalam kelas tersebut mempunyai karakteristik masing-masing yang terdefinisi dengan baik. Beberapa molekul-molekul monomer dapat digabungkan bersama membentuk sebuah entitas yang berukuran lebih besar, yang disebut macromolecule. Macromolecule dapat mempunyai informasi isi tertentu yang menarik dan sifat-sifat kimia tertentu. Berdasarkan skema di atas, monomer-monomer tertentu dalam macromolecule dari DNA dapat diperlakukan secara komputasi sebagai huruf-huruf dari alfabet, yang diletakkan dalam sebuah aturan yang telah diprogram sebelumnya untuk membawa pesan atau melakukan kerja di dalam sel. Proses yang diterangkan di atas terjadi pada tingkat molekul di dalam sel. Salah satu cara untuk mempelajari proses tersebut selain dengan mengamati dalam laboratorium biologi yang sangat khusus adalah dengan menggunakan Bioinformatika sesuai dengan definisi "klasik" yang telah disebutkan di atas.
2.1.5.2 Bioinformatika "baru"
Salah satu pencapaian besar dalam metode Bioinformatika adalah selesainya proyek pemetaan genom manusia (Human Genome Project). Selesainya proyek raksasa tersebut menyebabkan bentuk dan prioritas dari riset dan penerapan Bioinformatika berubah. Secara umum dapat dikatakan bahwa proyek tersebut membawa perubahan besar pada sistem hidup kita, sehingga sering disebutkan --terutama oleh ahli biologi-- bahwa kita saat ini berada di masa pascagenom. Selesainya proyek pemetaan genom manusia ini membawa beberapa perubahan bagi Bioinformatika, diantaranya:
Setelah memiliki beberapa genom yang utuh maka kita dapat mencari perbedaan dan persamaan di antara gen-gen dari spesies yang berbeda. Dari studi perbandingan antara gen-gen tersebut dapat ditarik kesimpulan tertentu mengenai spesies-spesies dan secara umum mengenai evolusi. Jenis cabang ilmu ini sering disebut sebagai perbandingan genom (comparative genomics). Sekarang ada teknologi yang didisain untuk mengukur jumlah relatif dari kopi/cetakan sebuah pesan genetik (level dari ekspresi genetik) pada beberapa tingkatan yang berbeda pada perkembangan atau penyakit atau pada jaringan yang berbeda. Teknologi tersebut, contohnya seperti DNA microarrays akan semakin penting. Akibat yang lain, secara langsung, adalah cara dalam skala besar untuk mengidentifikasi fungsi-fungsi dan keterkaitan dari gen (contohnya metode yeast twohybrid) akan semakin tumbuh secara signifikan dan bersamanya akan mengikuti Bioinformatika yang berkaitan langsung dengan kerja fungsi genom (functional genomics). Akan ada perubahan besar dalam penekanan dari gen itu sendiri ke hasil-hasil dari gen. Yang pada akhirnya akan menuntun ke: usaha untuk mengkatalogkan semua aktivitas dan karakteristik interaksi antara semua hasil-hasil dari gen (pada manusia) yang disebut proteomics; usaha untuk mengkristalisasi dan memprediksikan struktur-struktur dari semua protein (pada manusia) yang disebut structural genomics. Apa yang disebut orang sebagai research informatics atau medical informatics, manajemen dari semua data eksperimen biomedik yang berkaitan dengan molekul atau pasien tertentu --mulai dari spektroskop massal, hingga ke efek samping klinis-- akan berubah dari semula hanya merupakan kepentingan bagi mereka yang bekerja di perusahaan obat-obatan dan bagian TI Rumah Sakit akan menjadi jalur utama dari biologi molekul dan biologi sel, dan berubah jalur dari komersial dan klinikal ke arah akademis. Dari uraian di atas terlihat bahwa Bioinformatika sangat mempengaruhi kehidupan manusia, terutama untuk mencapai kehidupan yang lebih baik. Penggunaan komputer yang notabene merupakan salah satu keahlian utama dari orang yang bergerak dalam TI merupakan salah satu unsur utama dalam Bioinformatika, baik dalam Bioinformatika "klasik" maupun Bioinformatika "baru".
2.1.6 Cabang-cabang yang Terkait dengan Bioinformatika
Dari pengertian Bioinformatika baik yang klasik maupun baru, terlihat banyak terdapat cabang-cabang disiplin ilmu yang terkait dengan Bioinformatika --terutama karena Bioinformatika itu sendiri merupakan suatu bidang interdisipliner--. Hal tersebut menimbulkan banyak pilihan bagi orang yang ingin mendalami Bioinformatika. Di bawah ini akan disebutkan beberapa bidang yang terkait dengan Bioinformatika.
2.1.6.1 Biophysics
Biologi molekul sendiri merupakan pengembangan yang lahir dari biophysics. Biophysics adalah sebuah bidang interdisipliner yang mengaplikasikan teknik-teknik dari ilmu Fisika untuk memahami struktur dan fungsi biologi (British Biophysical Society). Sesuai dengan definisi di atas, bidang ini merupakan suatu bidang yang luas. Namun secara langsung disiplin ilmu ini terkait dengan Bioinformatika karena penggunaan teknik-teknik dari ilmu Fisika untuk memahami struktur membutuhkan penggunaan TI.
2.1.6.2 Computational Biology
Computational biology merupakan bagian dari Bioinformatika (dalam arti yang paling luas) yang paling dekat dengan bidang Biologi umum klasik. Fokus dari computational biology adalah gerak evolusi, populasi, dan biologi teoritis daripada biomedis dalam molekul dan sel. Tak dapat dielakkan bahwa Biologi Molekul cukup penting dalam computational biology, namun itu bukanlah inti dari disiplin ilmu ini.
Pada penerapan computational biology, model-model statistika untuk fenomena biologi lebih disukai dipakai dibandingkan dengan model sebenarnya. Dalam beberapa hal cara tersebut cukup baik mengingat pada kasus tertentu eksperimen langsung pada fenomena biologi cukup sulit. Tidak semua dari computational biology merupakan Bioinformatika, seperti contohnya Model Matematika bukan merupakan Bioinformatika, bahkan meskipun dikaitkan dengan masalah biologi.
2.1.6.3 Medical Informatics
Menurut Aamir Zakaria [ZAKARIA2004] Pengertian dari medical informatics adalah "sebuah disiplin ilmu yang baru yang didefinisikan sebagai pembelajaran, penemuan, dan implementasi dari struktur dan algoritma untuk meningkatkan komunikasi, pengertian dan manajemen informasi medis." Medical informatics lebih memperhatikan struktur dan algoritma untuk pengolahan data medis, dibandingkan dengan data itu sendiri. Disiplin ilmu ini, untuk alasan praktis, kemungkinan besar berkaitan dengan data-data yang didapatkan pada level biologi yang lebih "rumit" --yaitu informasi dari sistem-sistem superselular, tepat pada level populasi—di mana sebagian besar dari Bioinformatika lebih memperhatikan informasi dari sistem dan struktur biomolekul dan selular.
2.1.6.4 Cheminformatics
Cheminformatics adalah kombinasi dari sintesis kimia, penyaringan biologis, dan pendekatan data-mining yang digunakan untuk penemuan dan pengembangan obat (Cambridge Healthech Institute’s Sixth Annual Cheminformatics conference). Pengertian disiplin ilmu yang disebutkan di atas lebih merupakan identifikasi dari salah satu aktivitas yang paling populer dibandingkan dengan berbagai bidang studi yang mungkin ada di bawah bidang ini. Salah satu contoh penemuan obat yang paling sukses sepanjang sejarah adalah penisilin, dapat menggambarkan cara untuk menemukan dan mengembangkan obatobatan hingga sekarang --meskipun terlihat aneh--. Cara untuk menemukan dan mengembangkan obat adalah hasil dari kesempatan, observasi, dan banyak proses kimia yang intensif dan lambat.
Sampai beberapa waktu yang lalu, disain obat dianggap harus selalu menggunakan kerja yang intensif, proses uji dan gagal (trial-error process). Kemungkinan penggunaan TI untuk merencanakan secara cerdas dan dengan mengotomatiskan proses-proses yang terkait dengan sintesis kimiawi dari komponenkomponen pengobatan merupakan suatu prospek yang sangat menarik bagi ahli kimia dan ahli biokimia. Penghargaan untuk menghasilkan obat yang dapat dipasarkan secara lebih cepat sangatlah besar, sehingga target inilah yang merupakan inti dari cheminformatics. Ruang lingkup akademis dari cheminformatics ini sangat luas. Contoh bidang minatnya antara lain: Synthesis Planning, Reaction and Structure Retrieval, 3-D Structure Retrieval, Modelling, Computational Chemistry, Visualisation Tools and Utilities.
2.1.6.5 Genomics
Genomics adalah bidang ilmu yang ada sebelum selesainya sekuen genom, kecuali dalam bentuk yang paling kasar. Genomics adalah setiap usaha untuk menganalisa atau membandingkan seluruh komplemen genetik dari satu spesies atau lebih. Secara logis tentu saja mungkin untuk membandingkan genom-genom dengan membandingkan kurang lebih suatu himpunan bagian dari gen di dalam genom yang representatif.
2.1.6.6 Mathematical Biology
Mathematical biology lebih mudah dibedakan dengan Bioinformatika daripada computational biology dengan Bioinformatika. Mathematical biology juga menangani masalah-masalah biologi, namun metode yang digunakan untuk menangani masalah tersebut tidak perlu secara numerik dan tidak perlu diimplementasikan dalam software maupun hardware. Bahkan metode yang dipakai tidak perlu "menyelesaikan" masalah apapun; dalam mathematical biology bisa dianggap beralasan untuk mempublikasikan sebuah hasil yang hanya menyatakan bahwa suatu masalah biologi berada pada kelas umum tertentu.
Menurut Alex Kasman [KASMAN2004] Secara umum mathematical biology melingkupi semua ketertarikan teoritis yang tidak perlu merupakan sesuatu yang beralgoritma, dan tidak perlu dalam bentuk molekul, dan tidak perlu berguna dalam menganalisis data yang terkumpul.
2.1.6.7 Proteomics
Istilah proteomics pertama kali digunakan untuk menggambarkan himpunan dari protein-protein yang tersusun (encoded) oleh genom. Ilmu yang mempelajari proteome, yang disebut proteomics, pada saat ini tidak hanya memperhatikan semua protein di dalam sel yang diberikan, tetapi juga himpunan dari semua bentuk isoform dan modifikasi dari semua protein, interaksi diantaranya, deskripsi struktural dari proteinprotein dan kompleks-kompleks orde tingkat tinggi dari protein, dan mengenai masalah tersebut hampir semua pasca genom. Michael J. Dunn [DUNN2004], Pemimpin Redaksi dari Proteomics mendefiniskan kata "proteome" sebagai: "The PROTEin complement of the genOME".
Dan mendefinisikan proteomics berkaitan dengan: "studi kuantitatif dan kualitatif dari ekspresi gen di level dari protein-protein fungsional itu sendiri". Yaitu: "sebuah antarmuka antara biokimia protein dengan biologi molekul". Mengkarakterisasi sebanyak puluhan ribu protein-protein yang dinyatakan dalam sebuah tipe sel yang diberikan pada waktu tertentu --apakah untuk mengukur berat molekul atau nilai-nilai isoelektrik protein-protein tersebut-- melibatkan tempat penyimpanan dan perbandingan dari data yang memiliki jumlah yang sangat besar, tak terhindarkan lagi akan memerlukan Bioinformatika.
2.1.6.8 Pharmacogenomics
Pharmacogenomics adalah aplikasi dari pendekatan genomik dan teknologi pada identifikasi dari target-target obat. Contohnya meliputi menjaring semua genom untuk penerima yang potensial dengan menggunakan cara Bioinformatika, atau dengan menyelidiki bentuk pola dari ekspresi gen di dalam baik patogen maupun induk selama terjadinya infeksi, atau maupun dengan memeriksa karakteristik pola-pola ekspresi yang ditemukan dalam tumor atau contoh dari pasien untuk kepentingan diagnosa (kemungkinan untuk mengejar target potensial terapi kanker).
Istilah pharmacogenomics digunakan lebih untuk urusan yang lebih "trivial" — tetapi dapat diargumentasikan lebih berguna-- dari aplikasi pendekatan Bioinformatika pada pengkatalogan dan pemrosesan informasi yang berkaitan dengan ilmu Farmasi dan Genetika, untuk contohnya adalah pengumpulan informasi pasien dalam database.
2.1.6.9 Pharmacogenetics
Tiap individu mempunyai respon yang berbeda-beda terhadap berbagai pengaruh obat; sebagian ada yang positif, sebagian ada yang sedikit perubahan yang tampak pada kondisi mereka dan ada juga yang mendapatkan efek samping atau reaksi alergi. Sebagian dari reaksi-reaksi ini diketahui mempunyai dasar genetik. Pharmacogenetics adalah bagian dari pharmacogenomics yang menggunakan metode genomik/Bioinformatika untuk mengidentifikasi hubungan-hubungan genomik, contohnya SNP (Single Nucleotide Polymorphisms), karakteristik dari profil respons pasien tertentu dan menggunakan informasi-informasi tersebut untuk memberitahu administrasi dan pengembangan terapi pengobatan. Secara menakjubkan pendekatan tersebut telah digunakan untuk "menghidupkan kembali" obat-obatan yang sebelumnya dianggap tidak efektif, namun ternyata diketahui manjur pada sekelompok pasien tertentu. Disiplin ilmu ini juga dapat digunakan untuk mengoptimalkan dosis kemoterapi pada pasien-pasien tertentu. Gambaran dari sebagian bidang-bidang yang terkait dengan Bioinformatika di atas memperlihatkan bahwa Bioinformatika mempunyai ruang lingkup yang sangat luas dan mempunyai peran yang sangat besar dalam bidangnya. Bahkan pada bidang pelayanan kesehatan Bioinformatika menimbulkan disiplin ilmu baru yang menyebabkan peningkatan pelayanan kesehatan.
2.2 GENtle
2.2.1 Pengertian
Gentle adalah alat yang berguna bagi ahli biologi molekuler untuk menganalisa dan mengedit file urutan DNA. Penghapusan Invitrogens lisensi akademik free-of-biaya Vector NTI v11 telah memiliki dampak yang parah pada banyak laboratorium biologi molekuler yang telah datang untuk mengandalkan alat ini, yang menyebabkan vendor lock-in efek dan membuat marah banyak ahli biologi molekular.
2.2.2 Sejarah
perangkat lunak yang digunakan dalam pengembangan GENtle dan pelaksanaannya pada platform yang didukung. Sebagian besar adalah domain publik atau perangkat lunak gratis, tersedia di bawah lisensi seperti GNU General Public License (GPL). Sebuah daftar lisensi open source dapat ditemukan di http://www.opensource.org/. Untuk Windows dan Max OSX, lingkungan pengembangan terpadu (IDE) yang digunakan. Versi Linux dikompilasi langsung dari baris perintah, menggunakan autoconf / automake tool set.
Untuk pengembangan berbagai versi GENtle, platform masing-masing digunakan. Windows pembangunan berlangsung pada Windows 2000 dan XP, versi Mac dikembangkan di bawah OSX 10.4 pada PowerBook G4 (PowerPC), dan versi Linux disusun di bawah UBUNTU OS 6.06 LTS Drapper Drake.
2.3 Dev-CPP
Dev-Cpp atau Dev-C++ adalah IDE yang didistribusikan di bawah GNU General Public License alias gratis, bebas, enak dipakai fungsinya untuk pemrograman dalam C dan Cpp IDE ini ditulis dalam Delphi. Proyek ini diselenggarakan oleh SourceForge. Dev-Cpp pada awalnya dikembangkan oleh programmer Colin Laplace. Dev-Cpp secara eksklusif berjalan pada Microsoft Windows.
2.4 SQlite
SQLite merupakan sebuah sistem manajemen basisdata relasional yang bersifat ACID-compliant dan memiliki ukuran pustaka kode yang relatif kecil, ditulis dalam bahasa C. SQLite merupakan proyek yang bersifat public domain yang dikerjakan oleh D. Richard Hipp.
2.5 MySQL
MySQL adalah sebuah perangkat lunak sistem manajemen basis data SQL (bahasa Inggris: database management system) atau DBMS yang multithread, multi-user, dengan sekitar 6 juta instalasi di seluruh dunia. MySQL AB membuat MySQL tersedia sebagai perangkat lunak gratis dibawah lisensi GNU General Public License (GPL), tetapi mereka juga menjual dibawah lisensi komersial untuk kasus-kasus dimana penggunaannya tidak cocok dengan penggunaan GPL.
Tidak sama dengan proyek-proyek seperti Apache, dimana perangkat lunak dikembangkan oleh komunitas umum, dan hak cipta untuk kode sumber dimiliki oleh penulisnya masing-masing, MySQL dimiliki dan disponsori oleh sebuah perusahaan komersial Swedia MySQL AB, dimana memegang hak cipta hampir atas semua kode sumbernya. Kedua orang Swedia dan satu orang Finlandia yang mendirikan MySQL AB adalah: David Axmark, Allan Larsson, dan Michael "Monty" Widenius.
Tidak seperti pada paradigma client-server umumnya, Inti SQLite bukanlah sebuah sistem yang mandiri yang berkomunikasi dengan sebuah program, melainkan sebagai bagian integral dari sebuah program secara keseluruhan. Sehingga protokol komunikasi utama yang digunakan adalah melalui pemanggilan API secara langsung melalui bahasa pemrograman. Mekanisme seperti ini tentunya membawa keuntungan karena dapat mereduksi overhead, latency times, dan secara keseluruhan lebih sederhana. Seluruh elemen basisdata (definisi data, tabel, indeks, dan data) disimpan sebagai sebuah file. Kesederhanaan dari sisi disain tersebut bisa diraih dengan cara mengunci keseluruhan file basis data pada saat sebuah transaksi dimulai.
2.6 TinyXML
TinyXML adalah kumpulan dari C + + file yang terdiri dari parser XML, mengubah teks XML menjadi, representasi internal terstruktur seperti pohon. Saya menggunakan TinyXML di GENtle untuk mengurai file dalam format XML, misalnya, GenBankXML. Hal ini juga digunakan secara internal untuk beberapa deskripsi urutan (misalnya, keberpihakan) dalam database. TinyXML adalah perangkat lunak gratis di bawah lisensi zlib dan tersedia di http://www.grinninglizard.com/tinyxml/.
2.7 NSIS
Nullsoft Scriptable INSTALL SYSTEM (NSIS) adalah software untuk membuat installer Windows, yaitu, dikompresi, arsip self-extracting yang memberikan dialog instalasi, uncompress dan menyalin file termasuk ke lokasi yang dipilih, dan mendaftar aplikasi diinstal dengan registri Windows . Proses instalasi dapat didefinisikan meskipun bahasa scripting sederhana, maka nama itu. Saya memilih NSIS karena mudah digunakan bagi pengguna akhir, memiliki bahasa scripting yang kuat, dan mendukung kompresi LZMA, salah satu algoritma kompresi terbaik untuk file executable, menghasilkan faktor kompresi lebih dari 4 untuk file GENtle dan terkait. NSIS tersedia di bawah lisensi gratis tanpa biaya di bawah http://nsis.sourceforge.net/.
2.8 WxWidgets
WxWidgets (sebelumnya wxWindows) adalah toolkit cross-platform, yang memungkinkan saya untuk menulis kode yang akan mengkompilasi dan berjalan pada beberapa sistem operasi yang berbeda, asalkan hal ini terkait dengan perpustakaan masing-masing pada OS tersebut. Saya menggunakan wxWidgets pada Windows (WXMSW), Mac (WXMAC), dan Linux (wxGTK), meskipun tersedia untuk platform lebih. Meskipun namanya, wxWidgets tidak hanya menyediakan komponen visual seperti windows, menu, dan tombol, tetapi juga menawarkan akses platform-independen untuk sistem file dan jaringan, sampai ke array dan string yang kelas sendiri, yang telah terbukti berguna untuk dukungan Unicode mereka . WxWidgets tersedia di bawah lisensi LGPL di http://wxwidgets.org/.
2.9 Doxygen
Doxygen adalah sistem dokumentasi kode. Ia bekerja dengan menganalisis komentar khusus diformat dalam sumber program, dan menghasilkan dijelaskan, terstruktur, dan saling deskripsi fungsi global dan variabel, kelas, metode, dan variabel anggota. Sementara beberapa format output yang tersedia, saya hanya menggunakan output HTML karena fleksibilitasnya. Doxygen berlisensi di bawah GPL, dan tersedia di http://www.stack.nl/ ~ dimitri / doxygen /.
2.10 Online Resources and Services
2.10.1 BLAST
Berdasarkan protein atau urutan nukleotida, BASIC LOKAL ALIGNMENT SEARCH TOOL (BLAST) dapat digunakan untuk menemukan urutan yang sama dalam database. Dalam lembut, baik dapat dicari melalui NCBI antarmuka layanan web, menggunakan BLASTP untuk protein dan BLASTN untuk urutan nukleotida, masing-masing. Sebuah pencarian mengembalikan keselarasan preview asli dan urutan diduga. Yang terakhir dapat diambil sebagai urutan baru. The NCBI layanan BLAST tersedia di http://www.ncbi.nlm.nih.gov/blast/index.shtml.
2.10.2 PubMed
PubMed adalah database oleh US National Library of Medicine, dikenal berisi data kunci dari kebanyakan karya ilmiah yang ada. Lembut termasuk modul untuk query PubMed, menawarkan antarmuka yang lebih baik dibandingkan dengan layanan online. PubMed dapat ditemukan di http://www.pubmed.gov.
2.10.3 Web-based tools
Lembut menawarkan untuk menjalankan sebuah asam amino atau urutan nukleotida melalui berbagai alat online, termasuk fungsi untuk analisis dan prediksi struktur primer, topologi, motif dan fungsi, struktur sekunder, dan modifikasi posttranslational:
●Phobius and Poly-Phobius (transmembrane prediction) (http://phobius.cgb.ki.se)
●Motif scan (http://myhits.isb-sib.ch)
●P-val FPScan (http://umber.sbs.man.ac.uk)
●ELM (Functional site prediction) (http://elm.eu.org)
●Jpred (http://www.compbio.dundee.ac.uk)
●GOR and HNN (http://npsa-pbil.ibcp.fr)
●Phosphorylation states mW+pI (http://scansite.mit.edu)
●MitoProt II (http://ihg.gsf.de)
●Myristoylator (http://www.expasy.org)
●Sulfinator (http://www.expasy.org)
●SUMOplot (http://www.abgent.com)
●2ZIP (leucine zipper prediction) (http://2zip.molgen.mpg.de)
●TargetP (subcellular localization) (http://www.cbs.dtu.dk)
●DGPI (http://129.194.185.165/dgpi)
●SAPS (http://www.isrec.isb-sib.ch)
●NEBcutter (http://tools.neb.com)
●Nomad (http://tools.neb.com)
●MultAlin (http://prodes.toulouse.inra.fr)
●WebLogo (http://weblogo.berkeley.edu)
●Translate (http://www.expasy.org)
●PrePS (Prenylation) (http://mendel.imp.ac.at)
●PlasMapper (http://wishart.biology.ualberta.ca)
2.10.4 SourceForge
SourceForge adalah layanan online terkenal yang menawarkan hosting gratis dari proyek open source. Ini termasuk kode sumber sistem versi seperti CVS dan SVN, manajemen file dirilis, homepage proyek hosting, sistem pelacakan bug, dan forum diskusi. Untuk Lembut, saya telah membuat sebuah proyek SourceForge ("lembut-m") untuk memudahkan sinkronisasi suntingan antara platform pengembangan saya berbeda, dan untuk mengambil keuntungan dari versi melalui CVS, yang juga membantu dalam mendistribusikan up-to-date file sumber. Juga, ini akan memungkinkan untuk kolaborasi lebih programmer pembangunan lembut di masa depan. Halaman proyek tersedia di http://sourceforge.net/projects/gentle-m.
2.10.5 WikiBooks
Wikibooks adalah spin-off dari Wikipedia, ensiklopedia kolaboratif diedit dibawah lisensi bebas (Lisensi Dokumentasi Bebas GNU, GFDL). Demikian pula, Wikibooks bertujuan untuk menciptakan buku teks gratis, termasuk manual perangkat lunak. Untuk Lembut, saya telah beralih dari sistem bantuan yang terintegrasi ke petunjuk online di Wikibooks. Hal ini tidak hanya menyediakan up-to-date bantuan kepada pengguna, tetapi memungkinkan mereka untuk berkontribusi dan meningkatkan manual. Panduan ini tersedia di http://en.wikibooks.org/wiki/GENtle_manual.
BAB 3
PEMBAHASAN SOFTWARE
3.1 Instalasi
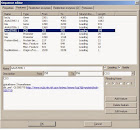
Untuk melakukan instalasi Gentle pada computer anda, pertama-tama yang harus dilakukan adalah menjalankan GENtleSetup.exe pada windows. Kemudian GENtle akan di instalasi pada direktori pilihan anda. Ada dua jenis database yang didukung oleh Gentle: berbasis file (sqlite) dan MySQL. Secara default, database local.db berbasis file diatur di direktori instalasi GENtle. Anda dapat (dan harus, jika keterbatasan akses mencegah Anda untuk bekerja dengan local.db) membuat database baru dan membaginya dengan pengguna lain dari GENtle dalam kelompok kerja Anda (jika Anda ingin). Manajemen database dapat ditemukan melalui menu "Tools / Manage database", di bawah tab "Database".
Catatan :
1. Hati-hati menggunakan local.db untuk pengembangan. Saya sangat menyarankan bahwa Anda tidak pernah memasukkan apa pun ke dalam local.db yang ada di direktori program GENtle dan jika, katakanlah, Anda perlu menginstal ulang software GENtle, atau Anda mendapatkan laptop baru, Anda mungkin mengkritik itu. Dan jika orang lain mengambil alih setelah itu, Anda juga bisa kehilangan program tersebut. Saya telah melihat perilaku aneh lain di mana local.db hilang, tetapi tidak dapat mengikat ke bawah untuk laporan bug.
2. GENtle menyalin blank.db dan local.db di [-username] / AppData / Lokal / VirtualStore / Program Files (x86) / GENtle,kemudian akan mengelola mereka. Salah satu Local.db yang ada terdapat di AppData / Lokal / VirtualStore / Program Files (x86) / GENtle. Jadi, Anda dapat menghapus satu di C :/ Program Files (x86) / GENtle , dan Anda tidak akan kehilangan itu. (Mungkin ini penyebab mengapa Magnus melakukannya dengan cara ini. Ia berusaha untuk mencegah kehilangan data.) Tapi, jika Anda mengganti komputer, local.db yang masih ada mungkin akan hilang. Jadi hal pertama yang harus Anda lakukan untuk pekerjaan serius adalah untuk membuat db baru Anda sendiri dan mengaturnya sebagai db default Anda. Dan memantau database lokal secara teratur untuk hal-hal tidak sengaja masuk ke database local tersebut.
3.2 Tools and Dialogs
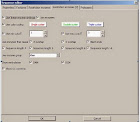
3.2.1 Ligation
Tampilan kotak dialog Ligation merupakan sarana untuk mengikat dua (atau lebih) fragmen DNA. Hal ini dapat dilakukan melalui Tools / Ligation atau Ctrl-L. List yang berada di sebelah kiri menunjukkan semua urutan DNA potensial untuk diikat. Beberapa di antaranya secara otomatis dipilih, tapi pilihan dapat diubah secara manual. List yang berada disebelah kanan menunjukkan kemungkinan produk hasil ikatan dari urutan yang dipilih. Beberapa produk melingkar akan ditampilkan dalam dua bentuk (AB dan BA), yang hanya berbeda secara visual. Produk yang dipilih akan dihasilkan sebagai urutan baru dengan mengklilk tombol Ligate.
3.2.2 Option
3.2.2.1 Global Setting
3.2.2.2 Enzyme settings
Disini pilihan enzim dapat dipilih. Ini dapat ditimpa untuk urutan individu dalam editor urutan, di mana ada tab identik dengan yang satu ini.
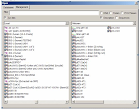
3.2.3 Database
Tampilan manajemen database GENtle adalah di mana urutan disimpan dan diambil. DNA dan asam amino urutan, primer, keberpihakan, dan proyek semua akan disimpan ke database, yang dapat bersifat lokal (untuk satu komputer saja) atau bersama (digunakan oleh kelompok kerja keseluruhan, lembaga, dll).
3.2.4 Import
Tampilan jendela import adalah standar tampilan"file yang terbuka". Hal ini dapat dipanggil melalui File / Import atau Ctrl-I. Beberapa file dapat dipilih untuk diimport berturut-turut. GENtle otomatis akan mencoba untuk menentukan jenis file, tetapi juga jenis file dapat dipilih secara manual. Format yang didukung termasuk: GenBank GenBank XML FASTA ABI/AB1 (format output sequencer populer) PDB (format 3D, impor sebagaimana dijelaskan urutan) Clone (program DOS, format proprietary) Banyak format urutan lain yang akan diimpor sebagai "urutan hanya", tanpa annnotations, fitur dll Comma-separated (CSV) dari berbagai mesin (HPLC / FPLC / fotometer) untuk grafik dan plot
3.2.5 Enter Sequance
Tampilan kotak dialog ini untuk memasukkan urutan secara manual dapat dipanggil melalui File / Masukkan urutan atau Ctrl-N. Selain urutan, yang akan diketik atau disisipkan ke dalam kotak teks besar, seseorang dapat memasukkan judul (nama) untuk urutan itu, dan memilih jenis a. Jenis yang tersedia adalah: DNA Urutan asam amino GenBank (GenBank) XML cat dasar Ketika memilih ponsel DNA, asam amino, atau primer, semua karakter non-urutan, seperti kosong dan nomor, secara otomatis dihapus.
3.2.6 Sequence Editor
Editor urutan memegang kunci untuk beberapa sifat berurutan. Ini terdiri dari beberapa tab, tergantung pada jenis urutan, yang bisa saja menjadi DNA atau asam amino.
3.2.6.1 Properties
Di sini, judul dan deskripsi dari urutan dapat diubah. Adapun deskripsi fitur, deskripsi urutan akan membuat referensi http untuk diklik. Untuk urutan DNA, ujung pada DNA dapat dimasukkan.
3.2.6.2 Features
Tab ini menampilkan daftar semua fitur dari urutan. Fitur dapat ditambahkan, diedit, dan dihapus. Sebagian besar pengaturan harus cukup jelas. Pengaturan membaca frame hanya bila tipe diatur ke "CDS" ("coding urutan"). Urutan terkemuka dibaca 5 ’→ 3’; paling depan dicentang, 3 ’→ 5’ Fitur edit akan memanggil tambahan "Edit fitur" dialog
Fill Color merupakan warna untuk fitur tersebut; itu akan memanggil tampilan dialog pilihan warna. Type display urutan menentukan bagaimana fitur yang digambarkan dalam peta urutan. Gunakan penetapan penomoran untuk asam amino pertama dari fitur; berguna jika fitur tersebut menandai bagian dari protein
3.2.6.3 Restriction Enzymes
Saat mengedit urutan DNA, dua tab tersedia dengan pengaturan untuk enzim. Yang pertama adalah identik dengan tampilan dialog manajemen enzim. Yang kedua adalah identik dengan tab pengaturan global enzim, tetapi berisi pengaturan untuk urutan ini saja. Secara default, opsi yang dinonaktifkan, dan opsi global yang digunakan. Dengan aktivasi opsi, pengaturan global diganti.
3.2.6.4 Proteases
Tab ini memiliki daftar protease yang tersedia. Potensi situs pembelahan untuk dipilih (diperiksa) protease ditunjukkan dalam urutan (tidak dalam peta DNA). Protease baru dapat ditambahkan mirip dengan contoh berikut: Contoh: "Thermolysin" Urutan untuk protease ini: "DE | AFILMV" Penjelasan: "Tidak D atau E", "cut", "maka A, F, I, L, M, atau V" Contoh: Proline-endopeptidase Urutan untuk protease ini: "! HKR, P | P" Penjelasan: "H, K, atau R", "maka P", "cut", "kemudian tidak P"
3.2.7 Restriction Assistant
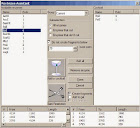
Restriction Assistant dapat dipanggil melalui menu Tools / Enzim Asisten, atau melalui klik dengan tombol tengah mouse di situs pembatasan dalam peta DNA. Untuk yang terakhir, enzim yang dipilih secara otomatis dipilih dalam daftar "Available enzymes" (kiri). Daftar ini tergantung pada pilihan "Group" dan "Subselection". Hal ini dapat diurutkan berdasarkan nama enzim atau jumlah pemotongan dengan mengklik judul kolom masing-masing. Untuk enzim yang dipilih, fragmen yang dihasilkan akan ditampilkan dalam daftar kiri bawah.
Daftar di sebelah kanan menunjukkan isi dari "restriction cocktail", enzim sudah dipilih untuk memotong. Yang dihasilkan fragmen untuk enzim ini bersama-sama akan ditampilkan dalam daftar kanan bawah. Enzim yang dipilih dalam daftar kiri dapat dimasukkan ke dalam koktail melalui Tambahkan ke koktail; semua enzim dari daftar kiri dapat ditambahkan sekaligus melalui Add semua. Sebuah enzim dapat dihapus dari koktail dengan memilihnya dalam daftar yang benar, kemudian melalui enzim Hapus.
Jangan membuat fragmen bawah ___ pasangan basa, saat dipilih, membatasi fragmen yang dihasilkan untuk ukuran minimum. Selesai keluar asisten pembatasan sambil menjaga perubahan Mage koktail, sedangkan Batal akan membatalkan semua perubahan yang dibuat. Mulai pembatasan (simbol gunting) akan memulai pembatasan simulasi. Hasil ini dapat dipengaruhi oleh beberapa pengaturan lebih lanjut:
• Buat fragmen akan menghasilkan urutan DNA yang sebenarnya dengan tumpul / ujung lengket mereka yang akan dihasilkan dari pencernaan dengan koktail. Pilihan ini adalah pra-dipilih.
• Tambahkan ke gel akan menambahkan fragmen ke gel virtual, bersama-sama dalam satu jalur.
• Masing-masing jalur akan mengubah yang berada di atasnya sehingga setiap enzim mendapat jalur sendiri.
• Pembatasan parsial akan menambahkan semua fragmen mungkin untuk jalur gel virtual, simulasi parsial (tidak lengkap) pembatasan. Opsi Satu jalur masing-masing tidak tersedia ketika pembatasan Partial diperiksa.
Restriction cocktail akan dipertahankan sehingga Anda dapat memotong DNA lain dengan kombinasi enzim, yang berguna untuk Ligation mendatang.
3.2.8 PCR troubleshoot dialog
Tampilan tools ini dapat dipanggil dari modul PCR and Primer Design. Modul ini untuk mengetes primer dalam modul untuk masalah-masalah khas. Catatan : bahwa ini tidak selalu daftar kesalahan, hanya indikator potensi masalah.
3.2.8.1 manual desain primer
Panduan primer designManual desain primer Serupa dengan modul asam amino, modul PCR didominasi oleh layar urutan, yang berisi DNA template dalam kedua 5 ’→ 3’ dan 3 ’→ 5’ notasi, fitur beranotasi, primer sebelah DNA, urutan DNA yang dihasilkan (produk PCR) dengan situs enzim restriksi, serta urutan yang dihasilkan asam amino (s) untuk kedua template dan DNA produk. Primer dapat diedit langsung di layar urutan; semua informasi urutan lainnya dilindungi (template DNA, fitur, dan asam amino) atau dihitung ulang tergantung pada template dan DNA primer (produk PCR, sehingga asam amino, situs restriksi). Primer dapat, selain melalui saran otomatis, diimpor atau ditambahkan secara manual. Primer dapat disimpan dalam database. Kedua fungsi anil setelah impor (untuk menempatkan primer di mana mereka akan anil kemungkinan besar) dan suhu leleh perhitungan didasarkan pada ujung 3 ’dari primer yang sesuai dengan urutan Template, karena ini adalah bagian yang paling penting bagi polimerase dan , dengan demikian, generasi produk PCR. Akibatnya, setiap nukleotida 5 ’dari mutasi pertama (dilihat dari ujung 3’) diabaikan untuk proses ini, yang dapat menyebabkan perhitungan suhu leleh sangat rendah. Ditingkatkan Algoritma sedang dikerjakan.
Layar urutan besar menunjukkan fitur dijelaskan, 5 ’primer (biru, ditandai pada latar belakang abu-abu), 5’ → 3 ’DNA template (hitam), urutan asam amino dari template (merah), 3’ → 5 ’DNA template (hitam ), 3 ’primer (urutan biru, tidak terlihat dalam gambar ini), sehingga PCR DNA produk (hijau) dengan situs restriksi (atas, red), dan menghasilkan asam amino (merah, bawah). Di bagian atas jendela, daftar primer terletak di sebelah kiri, dan menampilkan data kunci dari primer yang dipilih dalam daftar yang terletak di sebelah kanan.
Untuk menghindari variasi polimerase yang berbeda dan waktu perpanjangan, saya menambahkan parameter untuk panjang perpanjangan, yang disarankan secara otomatis, tergantung pada primer dan posisi anil mereka, tetapi dapat diubah secara manual. Daftar primer ditampilkan di atas layar urutan, dan untuk setiap primer, data kunci dapat dihitung dan ditampilkan. Seperti produk PCR dan informasi yang terkait, data ini diperbarui seketika saat mengedit primer, dan termasuk suhu leleh dihitung dengan tiga algoritma yang berbeda (metode GC, garam-disesuaikan, dan tetangga terdekat), panjang primer, GC isi dalam persen, dan yang paling mungkin diri anil primer dimer.
3.2.8.2 Mutagenesis Diam
Mutagenesis diam mutagenesis Diam adalah metode standar untuk memvalidasi keberhasilan PCR. A primer diubah dengan cara yang memperkenalkan situs pembatasan baru dalam produk PCR tanpa mengubah urutan asam amino.
Sebuah dialog khusus (Diam Mutagenesis Asisten, Gambar 8) memungkinkan untuk menemukan mutasi yang cocok untuk primer. Ini mencari mutasi diam parameter dengan set enzim, jumlah pemotongan, dan jumlah yang diperlukan pertukaran dasar. Atau, juga dapat mencari penghapusan situs pembatasan gantinya. Sebuah daftar menunjukkan mutasi yang sesuai, menunjukkan endonuklease bersangkutan, bursa dasar yang dibutuhkan, jumlah pemotongan, baik sebelum dan sesudah mutasi, urutan diubah primer, serta jumlah dan ukuran fragmen DNA setelah pembatasan dengan endonuklease masing. Pengguna dapat memilih salah satu saran, yang menyisipkan mutasi yang dipilih ke primer, dan memaksa tampilan situs pembatasan baru.
3.2.8.3 Optimasi Primer
Primer optimizationPrimer optimizationPrimer optimasi optimasi Primer Untuk meningkatkan primer dalam kisaran parameter, semua variasi primer selama 3 ’dan 5’ variasi, nilai minimum dan maksimum untuk kedua panjang primer dan suhu leleh dapat dihitung, ditimbang, dan ditampilkan. Pengguna dapat melihat data kunci dari salah satu primer baru, dan pilih salah satu dari mereka, memperbarui primer di layar urutan yang sesuai.
3.2.8.4 Pemecahan Masalah Assistant
Troubleshooting asisten asisten assistantTroubleshooting Masalah asisten asisten Primer mungkin menunjukkan berbagai masalah selama proses PCR yang sebenarnya. Untuk mencegah masalah, atau untuk mendiagnosa yang sudah ada, PCR Troubleshooting Asisten dapat memeriksa isi optimal GC, self-dan lintas-dimer, stabilitas, spesifisitas, mengulangi, dan GC berakhir. Sebagian besar algoritma didasarkan pada perhitungan energi bebas berdasarkan data dan formula dari Santalucia [19]. Beberapa algoritma yang digunakan selalu kembali hasilnya, dan beberapa dapat mengeluarkan peringatan bukan kesalahan yang signifikan. Dimana nilai-nilai melebihi batas yang telah ditetapkan yang menunjukkan kesalahan besar dengan primer atau kombinasi primer, peringatan ditekankan oleh semua huruf besar-huruf. 4.2.4.5 Virtual PCR Virtual PCR Setelah primer yang cukup dioptimalkan, urutan DNA yang dihasilkan dari proses PCR (atau urutan asam amino, tergantung pada frame membaca) dapat dihitung dan digunakan sebagai urutan baru. Semua fitur anotasi DNA template yang akan dipertahankan bila perlu.
3.2.9 Proteolysis Assistant
Proteolysis Assistant dapat dipanggil dari modul protein. Ini dapat membantu dengan memilih protease untuk proteolisis terbatas, pemisahan domain protein, dll Ada beberapa kotak daftar yang bergantung satu sama lain, sehingga mungkin memunculkan kebingungan pada awalnya.
Pertama dan terpenting, protein ini hampir dibelah oleh protease diperiksa dalam daftar Protease. Pemotongan dari semua protease dalam protein ini akan ditampilkan dalam daftar Cuts. Fragmen yang dihasilkan dari pemotongan ini akan ditampilkan dalam daftar Hasil fragmen, serta dalam simulasi Gel. Jika Anda tahu bahwa pemotongan tertentu tidak terjadi (misalnya, karena motif pengakuan dilindungi), Anda dapat membatalkan pilihan (uncheck) pemotongan ini dalam daftar Cuts. Jika Anda tahu bahwa fitur tertentu dijelaskan protein dilindungi dari protease, Anda dapat memeriksa mereka di Protect fitur daftar.
Jika Anda ingin memisahkan fitur dijelaskan (misalnya, domain) dari protein Anda melalui protease, Anda dapat memilih dua atau lebih fitur dalam daftar fragmen terpisah. Tergantung pada pilihan Anda dan keadaan Jumlah maksimum protease, daftar saran Cocktail akan diisi dengan protease (atau kombinasi dari itu) yang akan melakukan pekerjaan. Memilih salah satu dari "koktail", penjelasan yang lebih rinci akan ditampilkan di bawah daftar, dan protease masing-masing akan dipilih dalam daftar Protease.
Hasil akhir dari semua pengaturan ini adalah daftar fragmen Hasil. Anda dapat Urutkan fragmen dengan posisi cut, panjang fragmen, atau ukuran (berat). Anda dapat memeriksa fragmen individu, Semua atau tidak satupun dari mereka. Setelah OK, beberapa tindakan dapat dilakukan pada fragmen yang dipilih:
• Membuat fragmen yang akan dipilih akan membuat modul protein baru untuk setiap fragmen yang dipilih.
• Fragmen Anotasi terpilih akan menambahkan fitur untuk masing-masing fragmen yang dipilih dalam modul protein saat ini
• Mempertahankan protease akan menambahkan protease yang dipilih dalam daftar Protease ke modul protein saat ini
3.2.10 Project
Sebuah proyek di Gentle adalah kumpulan urutan yang dimiliki bersama-sama, bahkan jika mereka berada dalam database yang berbeda. Proyek dapat dimuat melalui File / Load Project atau F11 disimpan melalui File / Save Project atau F12 ditutup melalui File / Tutup Proyek Tergantung pada pilihan, proyek yang terakhir digunakan secara otomatis dibuka ketika Gentle dimulai. Proyek terdiri dari daftar urutan, bukan urutan sendiri. Jika urutan dinamai, dipindahkan atau dihapus, GENtle akan menampilkan peringatan setiap kali proyek yang memuat urutan yang dibuka. Untuk efisiensi penggunaan primer sequencing, seseorang dapat membuat sebuah proyek yang berisi semua primer sequencing yang tersedia, dan kemudian merujuk kepada proyek tersebut dalam tampilan Sequencing Primer.
3.2.11 Edit Primer Dialog
Fungsi dan tampilan ini membantu dalam mengoptimalkan primer. Untuk alasan itu, banyak varian dari primer yang dihasilkan dan dapat diperiksa.
Garis tengah dari tampilan menunjukkan variant primer saat ini ; rincian dari varian tersebut ditampilkan dalam kotak kanan atas. Dengan memilih OK, maka akan mengakhiri dialog tersebut, melakukan varian ke modul PCR. Cancel akan mengakhiri tampilan dan tidak mengubah modul PCR. Reset akan mengembalikan primer dalam dialog untuk varian dimulai dengan tampilan awalnya.
Daftar di bagian bawah tampilan,berisi daftar otomatis yang dihasilkan dari varian primer saat ini, diurutkan berdasarkan skor yang acak. The "region" dari varian dapat dipengaruhi oleh beberapa pengaturan pada kuartal kiri atas dialog. Pengaturan yang tersedia meliputi:
• Variasi dari 5’-akhir primer ke kanan dan ke kiri.
• Variasi dari 3’-akhir primer ke kanan dan ke kiri. • Panjang minimum dan maksimum primer.
• Suhu leleh minimum dan maksimum primer.
Setiap perubahan dari pengaturan ini akan memicu kalkulasi ulang kemungkinan variasi. Variasi ini kemudian dievaluasi dan ditampilkan dalam daftar di bagian bawah dialog. Double-klik salah satu varaiations akan mengubah variasi arus di garis tengah, dan display properties pada kuartal kanan atas dialog
Properties Display :
Disini akan ditampilkan :
1. Urutan primer dalam 5 ’→ 3’ orientasi
2. The ?H dan ?S nilai
3. Panjang dan GC isi
4. Suhu leleh, dihitung sesuai dengan metode Tetangga terdekat (biasanya hasil terbaik, tapi hanya untuk primer lagi)
5. Suhu leleh, dihitung sesuai dengan metode garam-disesuaikan (hasil medicore)
6. Suhu leleh, dihitung sesuai dengan metode GC (simplicistic)
7. Tertinggi skor self-anil (sewenang-wenang) dan tampilan yang anil
Peringatan: Menghitung suhu leleh primer adalah rumit. Jika salah satu dari tiga metode memberikan hasil yang sama sekali berbeda dibandingkan dengan dua lainnya, abaikan saja. Selain itu, suhu leleh hanya dihitung untuk 3’-akhir primer yang anneals dengan urutan!
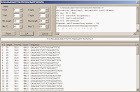
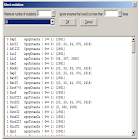
3.2.12 FIND
Tampilan Find pada DNA dan urutan asam amino dapat dipanggil melalui Ctrl-F atau Edit / Search. Fungsi find dapat digunakan untuk mencari dengan kategori :
• urutan saat ini
• nama fitur
• deskripsi fitur Dalam tampilan urutan DNA, juga terlihat di :
• urutan terbalik
• urutan asam amino yang diterjemahkan (s)
• nama enzim restriksi Pencarian akan dimulai secara otomatis setelah mengubah string pencarian, jika memenuhi tiga atau lebih karakter. Untuk permintaan pencarian yang lebih pendek, tombol “Search” harus diklik.
Single-klik pada hasil pencarian akan memilih dan menampilkan hasilnya dalam urutan. Sebuah klik ganda akan keluar dialog, dan membuka editor urutan untuk fitur, atau dari tampilan manajemen enzim untuk enzim restriksi.
3.2.13 Printing
Urutan dan Peta DNA dapat dicetak melalui menu konteks masing-masing atau menu File. Untuk urutan DNA, laporan dapat dicetak melalui laporan file / Print. Ini berisi peta DNA dan daftar fitur yang dijelaskan dalam urutan. Hal ini dapat berguna untuk gambaran rinci dari urutan,di mana urutan itu sendiri tidak diperlukan.
3.2.14 Enzyme Management
Editor enzim untuk manajemen enzim, baik secara global maupun per urutan DNA, dibagi menjadi tiga daftar:
• Daftar kelompok enzim (kanan atas)
• Daftar enzim dalam kelompok itu (kanan bawah)
• Daftar saat ini / enzim sementara (kiri)
Enzim dapat disalin ke / dihapus dari daftar kiri melalui <- Tambah dan Hapus -> tombol. Enzim dapat dihapus dari kelompok (kecuali All) melalui Delete dari grup, atau ditambahkan melalui enzim Baru. Double klik pada nama enzim dalam daftar menunjukkan tampilan propertis enzim.
Enzim dari daftar kiri dapat ditambahkan ke grup baru atau yang sudah ada melalui tombol masing-masing. Semua enzim dari kelompok dapat ditambahkan ke daftar kiri, dan kelompok dapat dihapus.
Anda dapat berbagi kelompok enzim dengan pengguna lain pada GENtle intranet anda melalui database umum bersama. Buat kelompok enzim sebagai "Database name:Enzyme group name ", dan database tersebut akan dapat digunakan oleh semua orang saat memulai GENtle. Perhatikan bahwa saat membuat kelompok enzim, gunakan nama database seperti yang muncul dalam instalasi lokal Anda (mungkin "Shared", "Shared0", "Shared (2)" dll).
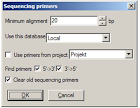
3.2.15 Sequencing Primers
Tampilan tools Sequencing Primers dapat menambahkan kemungkinan Sequencing Primer sebagai fitur untuk urutan DNA. Primer apa yang dapat ditambahkan bias dispesifikasikan sebagai brikut : • Minimum keselarasan (3 ’) dari primer ke urutan. Ini berarti anil yang tepat! • Database untuk mencari primer. Semua primer dari database yang akan dipertimbangkan. • Atau, gunakan semua primer yang merupakan bagian dari sebuah proyek di database tersebut. Dengan begitu, berbagai primer di seluruh database dapat ditentukan dalam sebuah proyek dan dianggap sebagai primer sequencing di sini. • Primer yang berjalan dalam 5 ’→ 3’ atau 3 ’→ 5’ arah.
Anda juga bias mendapatkan tampilan dialog untuk menghapus Sequencing Primer yang lama dari urutan. Hal ini juga dapat dilakukan secara manual melalui Sunting / Remove sequencing primer dalam modul DNA. CatatanSequencing primer, jika tidak dihapus, akan disimpan sebagai fitur bersama-sama dengan urutan; mereka masih bisa dihapus kapan saja. Sequencing Primer akan tampil sebagai fitur kuning, dimana warna kuning tergantung pada arah mereka. Fitur sequencing primer aktual hanya selama 3 ’anil primer, sehingga primer sebenarnya mungkin lebih lama dari fitur ke arah ujung 5’. Untuk rincian, lihat deskripsi fitur, yang berisi urutan primer asli, antara data lainnya.
3.2.16 Silent Mutagenesis
Fungsi dari tools ini untuk dapat menemukan enzim restriksi yang memotong dalam urutan DNA ditandai (menu konteks Seleksi / Tampilkan enzim yang memotong di sini dalam modul DNA). Hal ini juga dapat menemukan versi alternatif dari DNA yang akan diterjemahkan ke dalam urutan asam amino yang sama, tetapi mengandung situs restriksi baru (mutasi diam). Sebuah enzim yang dipilih / mutasi akan muncul dalam urutan (DNA atau primer, masing-masing) pada OK. Hasilnya dapat diubah dengan :
• perubahan enzim yang kelompok untuk mencari
• membatasi jumlah kali enzim dapat memotong di seluruh urutan
• membatasi jumlah mutasi yang diperlukan untuk situs restriksi untuk mewujudkan (modul PCR saja)
3.2.17 Automatic Annotation
Fitur Automatic Annotation dapat mencari database vektor standar (disertakan dengan paket GENtle), dan (opsional) a user-generated database, urutan fitur yang ditemukan dalam urutan DNA yang sedang dibuka. Fitur Diakui kemudian dijelaskan dalam urutan saat ini. Dipanggil melalui Edit / Auto-annotate atau F9, dialog terbuka, menawarkan berbagai pengaturan:
• Apakah atau tidak untuk mencari database vektor umum • Apakah atau tidak untuk menggunakan database user-generated (dan, jika demikian, yang satu)
• Apakah atau tidak untuk mengurangi jumlah fitur yang dihasilkan (disarankan, jika tidak, banyak fitur yang dijelaskan)
• Apakah atau tidak untuk menambahkan frame baca terbuka yang belum diakui sebagai fitur
3.3 Layout kode dan Database
3.3.1 Statistik kode
Pada September 2006, Gentle terdiri dari lebih dari 48.000 baris C + + kode yang ditulis sendiri, serta tambahan 49.000 baris asing C / C + + kode dan file header dari CLUSTALW, TinyXML, Ncoils, sqlite, UReadSeq, dan IPC. Ini tidak termasuk kode untuk perpustakaan digunakan (MySQL, sqlite, wxWidgets dan perpustakaan gambar yang terkait, serta standar C / C + + perpustakaan). Kode sendiri tersebar di 75 file sumber (tidak termasuk header). Secara total, ada sekitar 170 C + + kelas, termasuk kode asing. Karena jumlah kode dan kompleksitas yang terkandung di dalamnya, algoritma akan dijelaskan secara rinci di mana saja yang dianggap tepat. Untuk rincian implementasi, dokumentasi kode (→ 4.2.8) atau kode itu sendiri.
3.3.2 Database
Untuk menghindari duplikasi kode, MYSQL, SQLITE2 dan sqlite3 semua menggunakan struktur yang sama SQL (Figure2) dan perintah, seperti yang diterapkan oleh satu kelas. Panggilan database sebenarnya yang dilakukan dalam metode swasta, efektif dan selaput opaquing akses database dari sisa kode. Gentle dilengkapi dengan dua database: A "kosong" database yang berisi apa-apa selain daftar enzim restriksi dan protease, dan database yang berisi vektor umum untuk fungsi anotasi otomatis. Database sqlite3 baru diciptakan oleh
Figure 2: Sekema Database GENtle Nama tabel berwarna biru dan kuning menunjukan kunci sedangkan tanda panah mengindikasikan referensi nilai dari tabel lain. Menyalin database kosong, sementara database MYSQL baru diciptakan melalui serangkaian perintah SQL dalam file juga disertakan dengan Gentle.
Awalnya, SQLITE2 digunakan secara eksklusif, namun, ini menjadi masalah karena SQLITE2 tidak menyimpan data melebihi 1MB dalam ukuran. Dengan demikian, SQLITE2 termasuk untuk mundur kompatibilitas; Database SQLITE2 akan dikonversi ke sqlite3 on-the-fly jika ada upaya untuk menerobos hambatan 1MB oleh pengguna. Database baru dibuat secara eksklusif sebagai sqlite3 atau MYSQL. Semua database yang diakses melalui interface umum (Gambar 3). Data dapat disaring oleh kata kunci dan tipe data (DNA, sekuens asam amino, primer, keberpihakan). Kata kunci dapat dicari dalam nama entri, deskripsi, dan urutan. Data dapat dipindahkan atau disalin dari satu database ke yang lain dengan drag-and-drop, diganti atau dihapus. Sebagai fungsi keamanan terhadap sengaja Timpa entri, menyimpan entri diblokir jika ada sudah ada entri dengan urutan yang berbeda dengan nama itu dalam database.
3.3.3 Sequence Data
Secara internal, urutan data dari kedua asam amino DNA dan disimpan dalam satu kelas, TVector, yang berisi semua metode dasar untuk annotating, menganalisis dan memanipulasi urutan masing-masing. Sementara ini pada awalnya tampaknya melanggar makna dari model warisan kelas terhadap jenis kelas yang lebih spesifik, itu terbukti sangat berguna dalam praktek, karena kelas dapat bertindak sebagai tempat penyimpanan urutan universal.
3.3.4 Sequence Display
Sebuah kelas SequenceCanvas tunggal digunakan untuk menampilkan semua urutan dan informasi yang terkait, seperti situs restriksi, fitur, dan asam amino yang dihasilkan. Untuk itu, tampilan urutan terdiri dari beberapa baris dari berbagai jenis, dengan masing-masing jenis yang diwakili oleh kelas sendiri. Kelas baris ini semua anak-anak dari SeqBasic kelas, yang membawa satu set dasar variabel anggota dan metode. Garis urutan diatur setiap kali salah satu dari mereka berubah, dan sebaliknya ditampilkan dalam pengaturan terbaru mereka. Dalam kondisi tertentu, pengaturan cache posisi elemen garis, misalnya, dalam sequencer tampilan data. Biasanya, meskipun, posisi elemen dihitung pada gambar, yang dapat menghemat sejumlah besar memori, terutama ketika berhadapan dengan urutan yang lebih besar. Urutan menampilkan gulir vertikal secara default, simulasi tata letak halaman-seperti, tapi kebanyakan dari mereka dapat berubah menjadi mode horisontal bergulir. Semua menampilkan urutan dapat disesuaikan dalam ukuran font, dan dicetak, disalin, dan disimpan sebagai gambar bitmap.
3.3.5 Algoritma Runtime
Beberapa algoritma Gentle digunakan secara ekstensif, sehingga mempengaruhi keseluruhan kinerja. Runtime estimasi untuk ini adalah sebagai berikut:
● garis urutan menampilkan runtime adalah ? ? n ? dengan n adalah jumlah nukleotida atau asam amino. Pengecualian untuk ini adalah garis urutan untuk fitur, yang lebih buruk, dan dengan demikian dinonaktifkan secara default ketika menampilkan urutan yang sangat besar.
● Translation, juga, adalah ? ? n ? dengan n adalah jumlah nukleotida dalam urutan.
● pencarian situs Restriction adalah ? ? n × m ? dengan n adalah jumlah nukleotida dalam urutan, dan m adalah jumlah endonuklease restriksi untuk memeriksa. Namun, karena beberapa kondisi cutoff, runtime umumnya jauh lebih baik. Perilaku serupa diamati untuk deteksi situs membelah protease.
3.3.6 Update
Update GENtle secara otomatis mencari online ketika Gentle dimulai, menawarkan pengguna untuk men-download dan menjalankan update secara otomatis. Sebuah komentar singkat menyoroti perubahan, sehingga pengguna dapat memutuskan apakah update ini adalah sepadan dengan waktu dan bandwidth. Secara internal, program dan database versi ditangani secara terpisah. Setiap database membawa versi ditandai, menentukan versi basis data contoh Gentle mengakses harus memahami untuk mengoperasikan database tanpa masalah. Sebuah versi lama Gentle demikian dicegah dari sengaja merusak skema database baru. Jika kondisi demikian terjadi, pengguna akan diberitahu untuk memperbarui versi Gentle, dan contoh Gentle dapat mengakses database untuk membaca saja.
3.4 Modul
Gentle dibagi menjadi modul, yang menawarkan pemandangan dan fungsi cocok untuk konteks tertentu. Sementara banyak modul Gentle yang berbasis urutan, ada beberapa modul berdasarkan gambar atau data spreadsheet. Setiap modul terbuka tercantum dalam struktur pohon dikategorikan selalu terlihat di sisi kiri aplikasi GENtle, dan internal diwakili oleh kelas masing-masing. Semua kelas modul didasarkan pada kelas ChildBase induk, yang menawarkan metode dasar dan variabel anggota, termasuk antarmuka internal yang umum.
3.4.1 Urutan DNA
Salah satu modul inti GENtle adalah modul urutan DNA. Hal ini memungkinkan untuk tampilan, analisis, dan manipulasi dari urutan DNA.
Pohon Modul ini terletak di bagian paling kiri, DNA / plasmid peta di kanan atas, pohon sifat DNA di kiri atas, urutan DNA di bagian bawah. Sebagian ditandai urutan ditampilkan dalam warna abu-abu di kedua peta dan urutan. Peta menunjukkan isi GC, situs metilasi, dan frame baca terbuka. Kedua peta dan urutan acara situs enzim restriksi dan fitur dijelaskan. Urutan menunjukkan situs protease Faktor Xa. Layar urutan menunjukkan urutan DNA itu sendiri, dan opsional ’→ 5’ urutan DNA komplementer 3, fitur beranotasi, E. coli situs metilasi situs restriksi endonuklease, urutan asam amino yang dihasilkan dari DNA baik manual atau otomatis (fitur berbasis) reading frame (s) termasuk situs protease potensial. The tampilan peta di atas urutan menunjukkan sama tetapi untuk situs protease, dan tambahan dapat menampilkan frame terbuka membaca dan GC isinya, serta ujung lengket urutan linear, jika berlaku.
Peta ini dapat dicetak, disimpan, dan disalin ke clipboard sebagai gambar. DNA dapat ditandai di kedua display, dan diedit langsung di layar berurutan. Semua informasi yang tergantung pada DNA berubah seketika dengan DNA diedit. Dengan demikian, urutan asam amino yang dihasilkan berubah dengan urutan DNA yang telah berubah. Jika perubahan diperkenalkan memperkenalkan atau menghilangkan tempat pembelahan protease untuk protease yang dipilih, situs yang akan muncul atau menghilang, masing-masing, di layar berurutan. Demikian juga, situs restriksi baru diperkenalkan atau dihapus akan muncul atau menghilang sesuai.
Urutan dapat diedit, disalin atau dipotong secara manual, berubah (terbalik dan / atau urutan komplementer), dan helai dapat diekstraksi. Sebuah fungsi pencarian menemukan DNA membentang di kedua untai, situs enzim restriksi, asam amino membentang di semua frame membaca, dan fitur dijelaskan. Hasil dapat sementara disorot dalam urutan. Sekuens asam amino yang dapat diekstraksi melalui fitur atau diatur secara manual bingkai membaca dan urutan DNA menandai. Modul dapat menandai kopel siRNA potensial (→ 4.3.2.2), otomatis membubuhi keterangan urutan DNA dari kedua set termasuk vektor standar dan urutan sendiri dalam database, dan primer anil dari database yang bisa berfungsi sebagai sequencing primer ke urutan.
Pencarian BLAST (→ 4.4.1) dapat dilakukan untuk sekuens DNA atau asam amino. Informasi tambahan tentang urutan DNA, seperti nama dan ukuran, struktur pohon dengan fitur, daftar enzim restriksi, dan deskripsi diperlihatkan kiri layar peta. Seperti di sebagian besar modul, banyak fungsi dapat diterapkan untuk benda-benda seperti fitur dijelaskan, situs restriksi, dan (ditandai) urutan melalui menu konteks, yang akan muncul ketika mengklik pada objek seperti dengan tombol mouse sebelah kanan. Ini bekerja untuk semua jenis objek dalam pohon dan peta; menu konteks tampilan urutan terbatas untuk operasi urutan, fitur dan situs restriksi yang kadang-kadang sulit untuk membedakan dengan kursor mouse.
Modul urutan DNA juga mendukung tengah ("wheel") tombol mouse, jika tersedia, untuk memanggil Asisten Restriction di situs restriksi dan "mark dan menunjukkan" urutan fitur dijelaskan.
3.4.2 Sekuens Asam Amino
Modul asam amino urutan, struktur, mirip dengan modul urutan DNA (Gambar 5). Urutan dan fitur-fiturnya dijelaskan adalah inti dari layar berurutan. Pohon dan peta telah digantikan oleh fungsi pemilih ganda, menawarkan peta seperti skema urutan, halaman dengan catatan, set otomatis dihasilkan dari data kunci dari urutan, dan beberapa bidang fungsional sifat berurutan. Ini termasuk berat asam amino, titik isoelektrik, hidrofobik (→ 4.3.2.6), Chou-Fasman prediksi struktur sekunder (→ 4.3.2.5), dan prediksi melingkar-coil (→ 4.3.2.1). Versi kompresi dua terakhir, serta struktur asam amino molekul, juga dapat ditampilkan "sejalan", langsung di bawah urutan. Set data dan metode perhitungan untuk perhitungan data kunci diambil dari ExPASy.
Ini termasuk sejumlah asam amino, berat molekul (mW), diperkirakan titik isoelectic (pI), jumlah positif dan asam amino bermuatan negatif, jumlah rinci menyusun atom dan asam amino, diperkirakan paruh dalam organisme yang berbeda, dan grand rata dari hydropathicity (SAUS).
The major part of the module shows the amino acid sequence itself. Automatically numbered annotated features are shown above the sequence. Below the sequence, a simplified Chou-Fasman-plot [11] shows predicted α helices (red), β sheets (green), and turns (blue). Above the sequence display, the function selector is set to hydrophobicity, showing the respective plot in red.
Banyak tindakan, seperti mengedit dan mencari urutan, bekerja sama dengan modul urutan DNA juga, dengan adaptasi yang jelas (misalnya, tidak ada pencarian dalam bingkai membaca yang berbeda). Demikian juga, update fitur, data kunci, dan situs protease dilakukan seketika juga. Di antara fungsi untuk melakukan pada sekuens asam amino adalah pencarian BLAST (→ 4.4.1), perhitungan untuk analisis fotometri konsentrasi dan kemurnian, "backtranslating" ke dalam DNA menggunakan preferensi kodon standar atau spesies-spesifik, pola kalkulator isotop (→ 4.3. 2.3) untuk memprediksi data yang spektrometer massa untuk peptida pendek, dan Proteolisis Assistant.
3.5 Keberpihakan
Keberpihakan sangat penting untuk membandingkan urutan antara satu sama lain. Dalam Gentle, keberpihakan dapat dihasilkan lagi dari dua atau lebih sekuens menggunakan salah satu dari beberapa algoritma (→ 4.3.1), atau keberpihakan sudah dihasilkan dapat dibuka dari database atau diimpor dalam salah satu dari beberapa format file. Alignment dapat dioptimalkan secara manual dengan memasukkan dan menghapus kesenjangan. Urutan urutan dapat diubah, dan setiap urutan dapat menampilkan fitur aslinya. Gaya tampilan dapat diubah melalui kombinasi grafik warna standar dan pengeditan manual teks, latar belakang, dan warna perbatasan dan gaya, untuk lebih menekankan bagian-bagian penting dari keselarasan.
3.5.1 Phylip
Dari keselarasan dapat menciptakan sebuah pohon filogenetik, berdasarkan kesamaan dari dua sekuens masing-masing satu sama lain. Gentle dapat menampilkan pohon filogenetik yang dihasilkan oleh program PHYLIP software. Namun, program PHYLIP harus didownload dan diinstal secara terpisah; Aku tidak bisa memasukkannya ke dalam paket Gentle karena masalah perizinan. Gentle akan meminta lokasi biner program PHYLIP dan memperlakukannya secara transparan setelah itu, yaitu, lembut akan memanggil program PHYLIP dengan data pengguna dan menampilkan output tanpa memperhatikan pengguna panggilan dari program eksternal.
3.6 Sequencer Data
Saat ini, fungsi yang paling mesin sekuensing DNA berdasarkan metode terminasi rantai. Salah satu format yang paling sering digunakan untuk menyimpan kromatogram dari mesin ini adalah ABI (kadang-kadang AB1). Gentle native dapat membaca format ini, dan menampilkan kromatogram dalam modul sendiri. Filter impor untuk Format Standar Kromatogram (SCF) dan Ztr saat ini sedang dalam pembangunan. Urutan dapat diedit (hanya menimpa), disalin, dan disimpan dalam database. Aku telah menghilangkan penyimpanan data set lengkap sequencer, sebagai database sqlite mungkin kekacauan dari jumlah data (200-400 KByte per set data) dari waktu ke waktu. Ini mungkin berubah setelah sqlite dapat menyimpan data biner terkompresi. Keberpihakan data sequencer terhadap satu sama lain atau urutan lain dapat dilakukan secara manual atau melalui Sequencing Assistant, yang secara otomatis akan menentukan arah bacaan yang benar dari setiap kumpulan data sequencer, jika berlaku. Melalui mekanisme yang telah dijelaskan sebelumnya, pengguna dapat pergi dari ketidakcocokan keselarasan dengan nukleotida masing-masing dalam kromatogram secara manual membedakan antara urutan diubah atau salah pikiran dengan software sequencer.
3.7 Virtual Kloning
Virtual kloning bukan modul dalam dirinya sendiri, tetapi menjelaskan proses pemotongan virtual untai DNA dengan endonuklease restriksi, dan ligasi berikutnya dari beberapa DNA yang dihasilkan fragmen dengan urutan baru. Urutan DNA dapat dikenakan pencernaan virtual dengan endonuklease restriksi. Seorang asisten dialog (→ 5.2.12.2) memungkinkan untuk preview fragmen yang dihasilkan, dan generasi mereka sebagai urutan DNA baru dengan tumpul atau lengket berakhir. Pembelahan enzimatik juga dapat ditampilkan pada gel agarosa virtual. Proses sebaliknya, ligasi, dapat disimulasikan juga (→ 5.2.12.3). Dari daftar fragmen DNA, asisten dialog menunjukkan urutan DNA yang dihasilkan mungkin, baik linear dan melingkar, jika berlaku. Hasil yang dipilih dapat dihasilkan sebagai urutan DNA baru.
3.8 Virtual Gel
Untuk memvisualisasikan atau memvalidasi gel agarosa yang dihasilkan dari pembatasan endonuklease DNA, gel virtual dapat dihasilkan yang mensimulasikan elektroforesis. Asisten pembatasan dapat menambahkan pembatasan ke gel yang ada, atau membuat yang baru jika perlu. Jalur Gel dapat dibuat sebagai salah satu jalur per pembatasan endonuklease (enzim pencernaan tunggal), atau dengan semua hasil endonuklease pencernaan di jalur tunggal, baik lengkap maupun sebagian dicerna. Sebuah set diperpanjang tangga DNA tersedia untuk dijalankan terhadap sampel virtual. Gel ini dapat diubah dalam konsentrasi virtual. Semua label jalur yang diedit, dan konsentrasi agarosa dapat diubah. Gambar gel dapat disalin, dicetak, atau disimpan sebagai gambar. Tampilan gel, sengaja, tidak fotorealistik, untuk menghilangkan godaan bagi pengguna melewatkan elektroforesis gel aktual dan penerbitan in silico gel sebagai gantinya.
3.9 Antarmuka Web
Modul antarmuka web mengintegrasikan database pencarian di internet dengan lembut. Pencarian database dimulai dari dalam perangkat lunak, dan hasilnya ditampilkan dalam daftar untuk memilih dari.
Modul ini dapat mencari database dari National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/) untuk nukleotida dan asam amino urutan dengan nama. Entri urutan dikembalikan dapat dibuka melalui klik mouse ke urutan sepenuhnya dijelaskan. Atau, hasilnya dapat dibuka di situs web NCBI. Dalam cara yang sama, PubMed (→ 4.4.2) dapat dicari, menentukan kolom untuk kata kunci, penulis, dan berbagai tahun. Daftar Hasil penelitian menunjukkan preview dari judul paper, tanggal publikasi, jurnal, dan penulis. Hasil dapat klik-dibuka di browser web. Juga, BLAST (→ 4.4.1) mencari baik BLASTN (nukleotida) atau BLASTP (protein) dijalankan melalui antarmuka web (Gambar 11), meskipun mereka dipanggil dari DNA atau modul asam amino, masing-masing. Hasil BLAST ditunjukkan sebagai keselarasan sederhana asli dan pertandingan ditemukan bersama dengan nama pertandingan yang ditemukan serta "nilai E", yang menunjukkan kesamaan urutan. Sebuah klik mouse dapat membuka urutan pencocokan dijelaskan dalam modul urutan asam amino atau DNA baru, masing-masing.
3.10 Kromatogram
Modul penampil kromatogram adalah modul tidak-urutan berbasis. Hal ini dirancang untuk menampilkan data yang tercatat oleh HPLCs, FPLCs, fotometer, fluorimeters, dll Meskipun modul ini masih dalam tahap awal pengembangan, dapat membaca dan menampilkan beberapa format data, termasuk beberapa jenis photo-/fluorimeter, beberapa BioRad FPLCs , dan pasangan XY mentah sebagai comma-separated values (CSV). Layar auto-scaling dapat memperkecil, dan pilihan visual yang sedikit dapat diubah. Pandangan saat ini dapat disalin ke clipboard, dicetak, atau disimpan sebagai gambar. Direncanakan perbaikan dalam modul ini termasuk deteksi puncak otomatis dan integrasi.
3.11 Image Viewer
Modul penampil gambar adalah modul visualisasi lain. Tampaknya fungsi tidak mungkin untuk software biologi molekuler, itu lahir dari kebutuhan untuk mengakses gambar gel dan phosphoimager scan dalam format proprietary tua, kurang deskripsi apapun. Ini juga mendukung beberapa format gambar standar. Hal ini dapat menyimpan gambar dalam format standar terlepas dari format asli mereka, cetak, atau menyalinnya ke clipboard. Fitur yang direncanakan meliputi gambar lebih baku impor filter, serta deteksi band / plot dan analisis. 4.2.12 Kalkulator Kalkulator juga tidak berbasis-urutan. Setiap kalkulator adalah representasi berbasis spreadsheet dari fungsi yang sering terjadi dalam rutinitas sehari-hari laboratorium
3.12 Dialog dan Asisten
Ada beberapa dialog kecil dan besar dan asisten fungsi hadir dalam Gentle, mulai dari pilihan program global untuk permintaan nilai sederhana. Beberapa dari mereka telah dibahas; tiga lagi harus disebutkan secara rinci.
3.12.1 Urutan Editor
Editor urutan Editor urutan memungkinkan untuk mengedit kedua DNA dan urutan asam amino metadata, termasuk nama, deskripsi, dan protease yang aktif. Untuk urutan DNA, juga memungkinkan untuk penambahan endonuklease restriksi, baik secara manual maupun otomatis.
Manual manajemen pembatasan endonuklease memungkinkan pengguna memilih satu set endonuklease restriksi, yang luka dalam urutan akan ditampilkan jika berlaku. Untuk kenyamanan, kelompok enzim dapat didefinisikan dan diubah, misalnya, semua pembatasan endonuklease tersedia untuk kelompok kerja. Kelompok enzim ini dapat dibagi melalui database umum. Pemotongan restriksi endonuklease juga dapat ditampilkan untuk enzim sedemikian rupa sehingga sesuai dengan seperangkat aturan. Aturan-aturan ini dapat diatur secara global melalui dialog pilihan program (tidak terlihat), atau per urutan, mengesampingkan pengaturan global. Aturan mencakup batas jumlah minimal dan maksimal dari pemotongan enzim dalam urutan, jenis tumpang tindih daun dipotong, panjang urutan pengakuan, dan pilihan enzim dari kelompok enzim seperti yang disebutkan di atas. Selanjutnya, berisi opsi tampilan tentang gaya dan warna-coding dari enzim, serta opsi tampilan untuk metilasi (DAM dan DCM) dan isi GC urutan.
3.12.2 Asisten Restriction
Pembatasan asisten memungkinkan untuk mensimulasikan efek dari endonuklease restriksi pada urutan DNA. Enzim dari daftar yang dihasilkan dari kelompok tersebut dan sifat pemotongan dapat dikombinasikan untuk pembatasan "cocktail". Fragmen yang dihasilkan dari pencernaan dengan enzim tunggal atau koktail ditunjukkan dalam daftar masing-masing.
Hasil fragmen DNA dapat dihasilkan sebagai modul urutan DNA baru, opsional membatasi penciptaan fragmen yang sangat singkat. Sebuah gel virtual (→ 5.2.7) dapat dihasilkan untuk fragmen dari masing-masing enzim secara terpisah atau bersama-sama, opsional untuk digestions parsial juga. Urutan yang dihasilkan akan diurutkan di pohon utama di bawah "Fragmen DNA" untuk pemisahan yang lebih baik dari urutan sumber, tetapi tidak berbeda sebaliknya dari urutan DNA lain. Mereka akan diambil dari database di bawah judul DNA normal. 5.2.12.3 asisten Ligasi Asisten ligasi dapat mensimulasikan ligasi tunggal atau multipoint. Dari semua urutan non-melingkar, asisten berjalan melalui ligations mungkin dan menampilkan produk potensial. Urutan dan produk dapat dipilih oleh pengguna, dan produk dapat dihasilkan sebagai urutan baru.
BAB 4
STUDI KASUS
4.1 Program-program Bioinformatika
Sehari-harinya bionformatika dikerjakan dengan menggunakan program pencari sekuen (sequence search) seperti BLAST, program analisa sekuen (sequence analysis) seperti EMBOSS dan paket Staden, program prediksi struktur seperti THREADER atau PHD atau program imaging/modelling seperti RasMol dan WHATIF. Contoh-contoh di atas memperlihatkan bahwa telah banyak program pendukung yang mudah di akses dan dipelajari untuk menggunakan Bioinformatika
4.2 Teknologi Bioinformatika Secara Umum
Pada saat ini banyak pekerjaan Bioinformatika berkaitan dengan teknologi database. Penggunaan database ini meliputi baik tempat penyimpanan database "umum" seperti GenBank atau PDB maupun database "pribadi", seperti yang digunakan oleh grup riset yang terlibat dalam proyek pemetaan gen atau database yang dimiliki oleh perusahaan-perusahaan bioteknologi. Konsumen dari data Bioinformatika menggunakan platform jenis komputer dalam kisaran: mulai dari mesin UNIX yang lebih canggih dan kuat yang dimiliki oleh pengembang dan kolektor hingga ke mesin Mac yang lebih bersahabat yang sering ditemukan menempati laboratorium ahli biologi yang tidak suka komputer. Database dari sekuen data yang ada dapat digunakan untuk mengidentifikasi homolog pada molekul baru yang telah dikuatkan dan disekuenkan di laboratorium. Dari satu nenek moyang mempunyai sifat-sifat yang sama, atau homology, dapat menjadi indikator yang sangat kuat di dalam Bioinformatika.
Setelah informasi dari database diperoleh, langkah berikutnya adalah menganalisa data. Pencarian database umumnya berdasarkan pada hasil alignment / pensejajaran sekuen, baik sekuen DNA maupun protein. Kegunaan dari pencarian ini adalah ketika mendapatkan suatu sekuen DNA/protein yang belum diketahui fungsinya maka dengan membandingkannya dengan yang ada dalam database bisa diperkirakan fungsi daripadanya. Salah satu perangkat lunak pencari database yang paling berhasil dan bisa dikatakan menjadi standar sekarang adalah BLAST (Basic Local Alignment Search Tool) yang merupakan program pencarian kesamaan yang didisain untuk mengeksplorasi semua database sekuen yang diminta, baik itu berupa DNA atau protein. Program BLAST juga dapat digunakan untuk mendeteksi hubungan di antara sekuen yang hanya berbagi daerah tertentu yang memiliki kesamaan. Di bawah ini diberikan contoh beberapa alamat situs yang berguna untuk bidang biologi molekul dan genetika:
Data yang memerlukan analisa Bioinformatika dan mendapat banyak perhatian saat ini adalah data hasil DNA chip. Dengan perangkat ini dapat diketahui kuantitas dan kualitas transkripsi satu gen sehingga bisa menunjukkan gen-gen apa saja yang aktif terhadap perlakuan tertentu, misalnya timbulnya kanker, dan lain-lain.
4.3 Kondisi Bioinformatika di Indonesia
Di Indonesia, Bioinformatika masih belum dikenal oleh masyarakat luas. Hal ini dapat dimaklumi karena penggunaan komputer sebagai alat bantu belum merupakan budaya. Bahkan di kalangan peneliti sendiri, barangkali hanya para peneliti biologi molekul yang sedikit banyak mengikuti perkembangannya karena keharusan menggunakan perangkat-perangkat Bioinformatika untuk analisa data. Sementara di kalangan TI masih kurang mendapat perhatian. Ketersediaan database dasar (DNA, protein) yang bersifat terbuka/gratis merupakan peluang besar untuk menggali informasi berharga daripadanya. Database genom manusia sudah disepakati akan bersifat terbuka untuk seluruh kalangan, sehingga dapat digali/diketahui kandidat-kandidat gen yang memiliki potensi kedokteran/farmasi. Dari sinilah Indonesia dapat ikut berperan mengembangkan Bioinformatika. Kerjasama antara peneliti bioteknologi yang memahami makna biologis data tersebut dengan praktisi TI seperti programmer, dan sebagainya akan sangat berperan dalam kemajuan Bioinformatika Indonesia nantinya.
4.4 Penerapan Bioinformatika di Indonesia
Sebagai kajian yang masih baru, Indonesia seharusnya berperan aktif dalam mengembangkan Bioinformatika ini. Paling tidak, sebagai tempat tinggal lebih dari 300 suku bangsa yang berbeda akan menjadi sumber genom, karena besarnya variasi genetiknya. Belum lagi variasi species flora maupun fauna yang berlimpah. Memang ada sejumlah pakar yang telah mengikuti perkembangan Bioinformatika ini, misalnya para peneliti dalam Lembaga Biologi Molekul Eijkman. Mereka cukup berperan aktif dalam memanfaatkan kajian Bioinformatika. Bahkan, lembaga ini telah memberikan beberapa sumbangan cukup berarti, antara lain:
4.4.1 Deteksi Kelainan Janin
Lembaga Biologi Molekul Eijkman bekerja sama dengan Bagian Obstetri dan Ginekologi Fakultas Kedokteran Universitas Indonesia dan Rumah Sakit Cipto Mangunkusumo sejak November 2001 mengembangkan klinik genetik untuk mendeteksi secara dini sejumlah penyakit genetik yang menimbulkan gangguan pertumbuhan fisik maupun retardasi mental seperti antara lain, talasemia dan sindroma down. Kelainan ini bisa diperiksa sejak janin masih berusia beberapa minggu. Talasemia adalah penyakit keturunan di mana tubuh kekurangan salah satu zat pembentuk hemoglobin (Hb) sehingga mengalami anemia berat dan perlu transfusi darah seumur hidup. Sedangkan sindroma down adalah kelebihan jumlah untaian di kromosom 21 sehingga anak tumbuh dengan retardasi mental, kelainan jantung, pendengaran dan penglihatan buruk, otot lemah serta kecenderungan menderita kanker sel darah putih (leukemia).
Dengan mengetahui sejak dini, pasangan yang hendak menikah, atau pasangan yang salah satunya membawa kelainan kromosom, atau pasangan yang mempunyai anak yang menderita kelainan kromosom, atau penderita kelainan kromosom yang sedang hamil, atau ibu yang hamil di usia tua bisa memeriksakan diri dan janin untuk memastikan apakah janin yang dikandung akan menderita kelainan kromosom atau tidak, sehingga mempunyai kesempatan untuk mempertimbangkan apakah kehamilan akan diteruskan atau tidak setelah mendapat konseling genetik tentang berbagai kemungkinan yang akan terjadi. Di bidang talasemia, Eijkman telah memiliki katalog 20 mutasi yang mendasari talasemia beta di Indonesia, 10 di antaranya sering terjadi. Lembaga ini juga mempunyai informasi cukup mengenai spektrum mutasi di berbagai suku bangsa yang sangat bervariasi. Talasemia merupakan penyakit genetik terbanyak di dunia termasuk di Indonesia.
4.4.2 Pengembangan Vaksin Hepatitis B Rekombinan
Lembaga Biologi Molekul Eijkman bekerja sama dengan PT Bio Farma (BUMN Departemen Kesehatan yang memproduksi vaksin) sejak tahun 1999 mengembangkan vaksin Hepatitis B rekombinan, yaitu vaksin yang dibuat lewat rekayasa genetika. Selain itu Lembaga Eijkman juga bekerja sama dengan PT Diagnosia Dipobiotek untuk mengembangkan kit diagnostik.
4.4.3 Meringankan Kelumpuhan dengan Rekayasa RNA
Kasus kelumpuhan distrofi (Duchenne Muscular Dystrophy) yang menurun kini dapat dikurangi tingkat keparahannya dengan terapi gen. Kelumpuhan ini akibat ketidaknormalan gen distrofin pada kromosom X sehingga hanya diderita anak laki-laki. Diperkirakan satu dari 3.500 pria di dunia mengalami kelainan ini. Dengan memperbaiki susunan ekson atau bagian penyusun RNA gen tersebut pada hewan percobaan tikus, terbukti mengurangi tingkat kelumpuhan saat pertumbuhannya menjadi dewasa. Gen distrofin pada kasus kelumpuhan paling sering disebabkan oleh delesi atau hilangnya beberapa ekson pada gen tersebut.
Normalnya pada gen atau DNA distrofin terdapat 78 ekson. Diperkirakan 65 persen pasien penderita DMD mengalami delesi dalam jumlah besar dalam gen distrofinnya. Kasus kelumpuhan ini dimulai pada otot prosima seperti pangkal paha dan betis. Dengan bertambahnya usia kelumpuhan akan meluas pada bagian otot lainnya hingga ke leher. Karena itu dalam kasus kelumpuhan yang berlanjut dapat berakibat kematian. Teknologi rekayasa RNA seperti proses penyambungan (slicing) ekson dalam satu rangkaian terbukti dapat mengoreksi mutasi DMD. Bila bagian ekson yang masih ada disambung atau disusun ulang, terjadi perubahan asam amino yang membentuk protein. Molekul RNA mampu mengenali molekul RNA lainnya dan melekat dengannya.
4.4.4 Bioteknologi Dalam Bidang Pertanian
Bioteknologi telah diterapkan secara luas dalam bidang pertanian, antara lain yaitu: Pupuk Hayati (biofertiliser) yaitu suatu bahan yang berasal dari jasad hidup, khususnya mikrobia yang digunakan untuk meningkatkan kuantitas dan kualitas produksi tanaman. Kultur in vitro, yaitu pembiakan tanaman dengan menggunakan bagian tanaman yang ditumbuhkan pada media bernutrisi dalam kondisi aseptik. Kultur in vitro memungkinkan perbanyakan tanaman secara massal dalam waktu yang singkat. Teknologi DNA Rekombinaan, pengembangan tanaman transgenik, misalnya galur tanaman transgenik yang membawa gen cry dari Bacillus thuringiensis untuk pengendalian hama.
BAB 5
PENUTUP
5.1 Kesimpulan
Bioinformatika adalah teknologi pengumpulan, penyimpanan, analisis, interpretasi, penyebaran dan aplikasi dari data-data biologi molekul. Dengan Bioinformatika, data-data yang dihasilkan dari proyek genom dapat disimpan dengan teratur dalam waktu yang singkat dengan tingkat akurasi yang tinggi serta sekaligus dianalisa dengan program-program yang dibuat untuk tujuan tertentu.
Software GENtle tidak hanya berguna bagi ahli biologi molekuler untuk menganalisa dan mengedit file urutan DNA tetapi juga sangat membantu dan saling menunjang sehingga bermanfaat untuk kepentingan manusia. Bidang yang terkait dengan Bioinformatika diantaranya adalah Biophysics, Computational Biology, Medical Informatics, Cheminformatics, Genomics, Mathematical Biology, Proteomics, Pharmacogenomics.
Demikianlah yang dapat kami sampaikan mengenai materi yang menjadi bahasan dalam buku ini, tentunya banyak kekurangan dan kelemahan kerena terbatasnya pengetahuan kurangnya rujukan atau referensi yang kami peroleh hubungannya dengan materi ini kami banyak berharap kepada para pembaca yang budiman memberikan kritik saran yang membangun kepada kami demi sempurnanya buku ini. Semoga buku ini dapat bermanfaat dan membantu bagi penulis dan pembaca.
Bibliography
[1] [BIOINFORMATICS2004] BioInformatics.org: The Open-Access Institute, http://bioinformatics.org per 20 Juni 2014
[2] [DUNN2004] Majalah Proteonomics, http://www.wiley.co.uk/wileychi/genomics/proteomics.html/ per 20 Juni 2014
[3] [KASMAN2004] Situs Alex Kasman di College of Charleston, http://math.cofc.edu/faculty/kasman/ per 20 Juni 2014
[4] Poster presentation at the BioPerspectives congress in Wiesbaden, Germany (May 2005)
[5] [TEKAIA2004] Situs Institut Pasteur, http://www.pasteur.fr/externe per 20 Januari 2004 [ZAKARIA2004] Medical Informatics FAQ, http://www.faqs.org/faqs/medicalinformatics- faq/ per 20 Juni 2014
[6] UTAMA2003] Utama, Andi (2003), Peranan Bioinformatika dalam Dunia Kedokteran, http://ikc.vlsm.org/populer/andi-bioinformatika.php per 20 Juni 2014.
[7] [WITARTO2003] Witarto, Arief B. (2003), BIOINFORMATIKA: Mengawinkan Teknologi Informasi dengan Bioteknologi. Trendnya di Dunia dan Prospeknya di Indonesia
[8] [ZAKARIA2004] Medical Informatics FAQ, http://www.faqs.org/faqs/medicalinformatics- faq/ per 20 Juni 2014
T0:
Job Description

Milestones

Minutes of Meeting